
Mines Physique 1 PSI 2002
| Thème de l'épreuve | Transition de phase et transition ordre-désordre |
| Principaux outils utilisés | changements d'état, dérivations partielles et principes thermodynamiques, probabilités sur réseau cristallin |
| Mots clefs | équation de Van der Waals, point critique, transition de phase |
Corrigé
:👈 gratuite pour tous les corrigés si tu crées un compte
👈 l'accès aux indications de tous les corrigés ne coûte que 1 € ⬅ clique ici
👈 gratuite pour tous les corrigés si tu crées un compte
- - - - - - - - - - - - - - - - - - - - - - -
👈 gratuite pour ce corrigé si tu crées un compte
- - - - - - - - -
Énoncé complet
(télécharger le PDF)




Rapport du jury
(télécharger le PDF)


Énoncé obtenu par reconnaissance optique des caractères
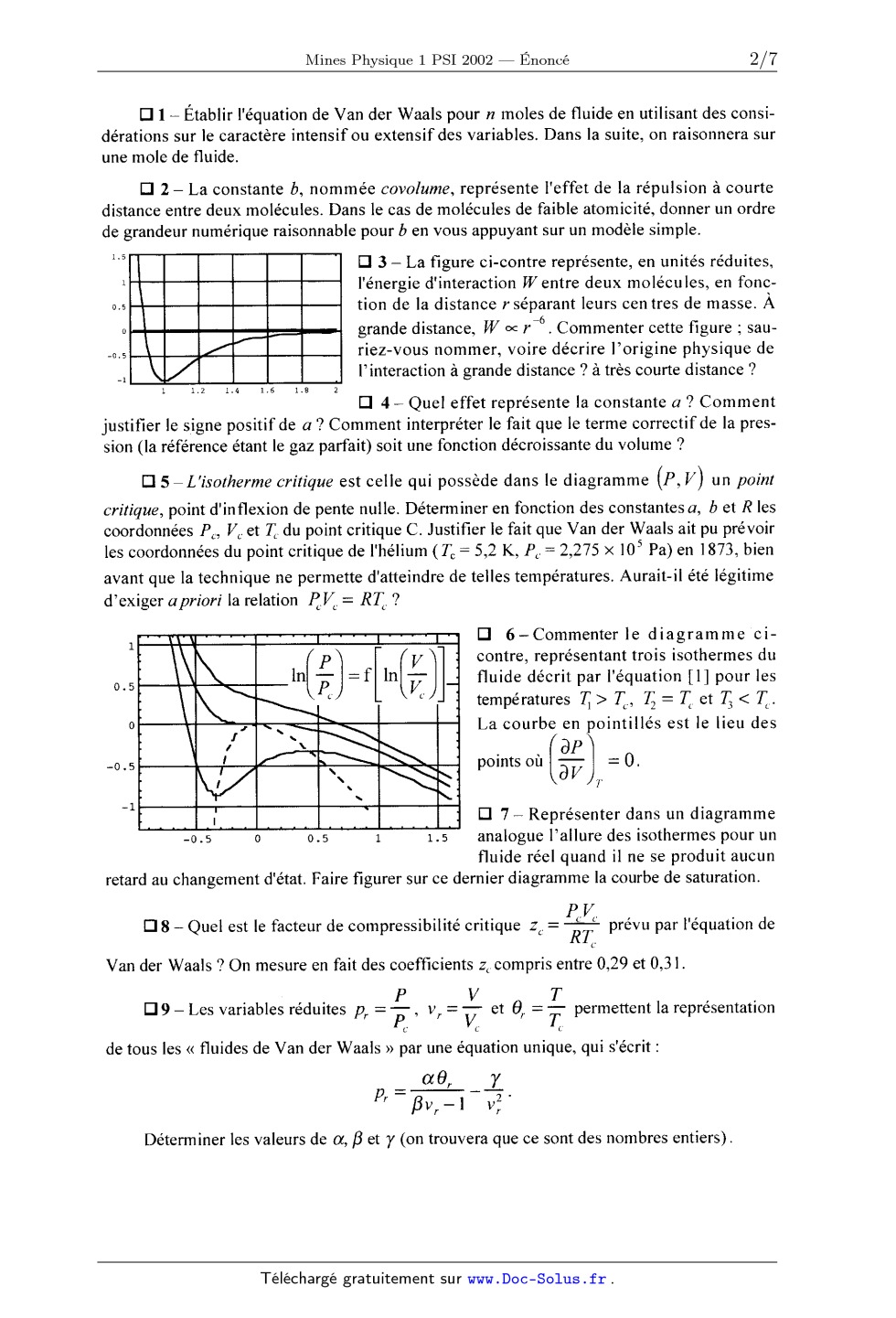
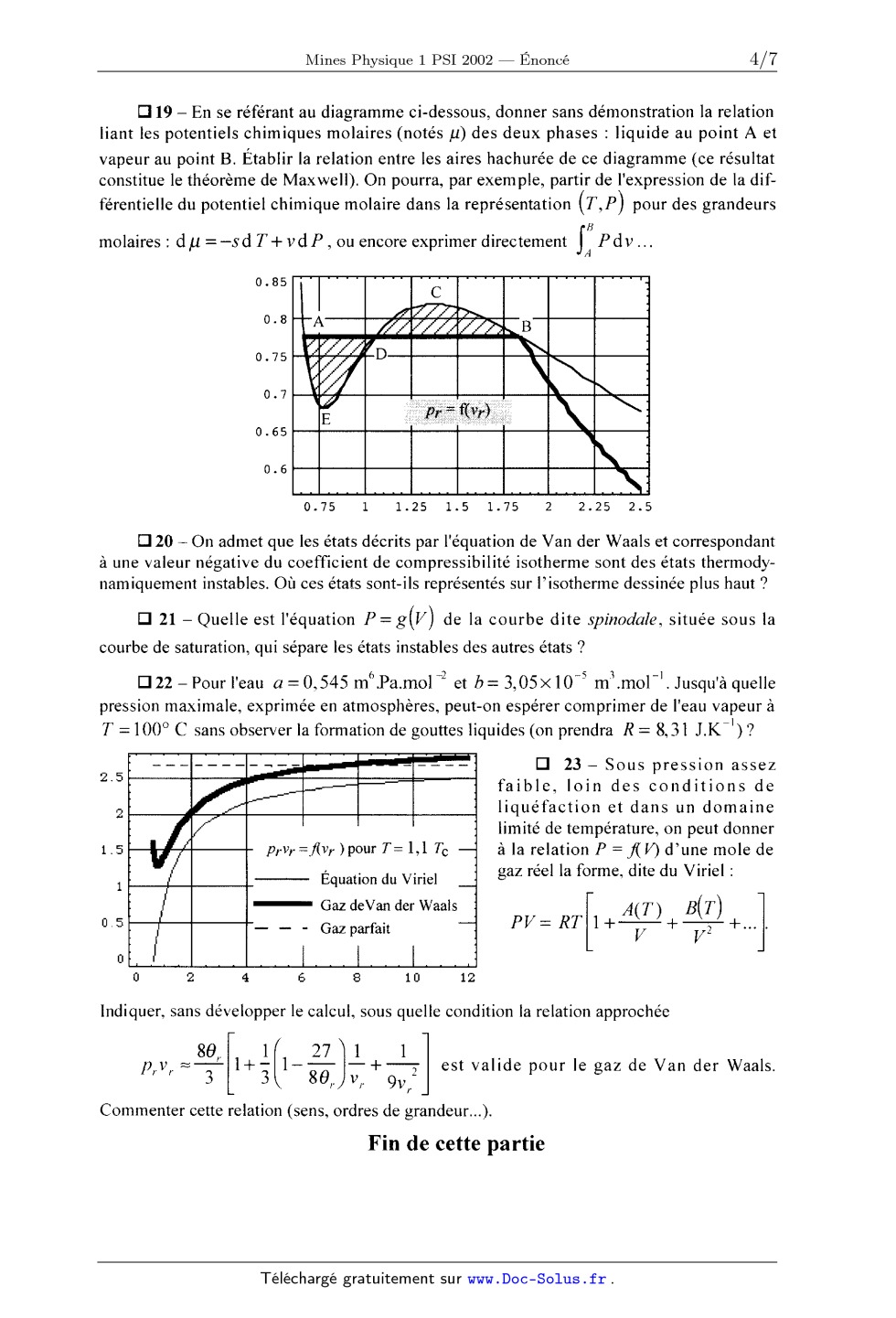
J. 2160 ECOLE NATIONALE DES PONTS ET CHAUSSÉES, ÉCOLES NATIONALES SUPÉRIEURES DE L'AÉRONAUTIQUE ET DE L'ESPACE DE TECHNIQUES AVANCEES, DES TÉLÉCOMMUNICATIONS, DES MINES DE PARIS, DES MINES DE SAINT-ÉTIENNE, DES MINES DE NANCY, DES TÉLÉCOMMUNICATIONS DE BRETAGNE ECOLE POLYTECHNIQUE (FILIÈRE TSI) CONCOURS D'ADMISSION 2002 PREMIÈRE ÉPREUVE DE PHYSIQUE Filière PSI (Durée de l'épreuve : 3 heures ; l'usage de la calculatrice est autorisé) Sujet mis à disposition des concours : Cycle international, ENSTIM, INT, TPE-EIVP Les candidats sont priés de mentionner de façon apparente sur la première page de la copie : Physique I -- Filière PSI L'én0nce' de cette épreuve, particulière aux candidats de la filière PSI, comporte 7 pages. 0 Si, au cours de l'épreuve, un candidat repère ce qui lui semble être une erreur d'énoncé, il le signale sur sa copie et poursuit sa composition en expliquant les raisons des initiatives qu'il est amené à prendre. 0 Tout résultat fourni dans l'énoncé peut être utilisé pour les questions ultérieures, même S'il n'a pas été démontré. . Il ne faudra pas hésiter à formuler tout commentaire qui vous semblera pertinent, même lors-- que l'énoncé ne le demande pas explicitement. Le barème tiendra compte de ces initiatives ainsi que des qualités de rédaction de la copie. ASPECTS DES TRAN SITIONS Les deux parties proposées dans cette épreuve sont indépendantes. La première partie étudie l'équilibre d'un fluide décrit par l'équation de VAN DER WAALS et la transition d'état liquide- vapeur. La transition ordre-désordre dans un alliage solide est étudiée en seconde partie grâce à la notion de paramètre d'ordre. 1 - Forces et limites de l'équation d'état de Van der Waals. À la différence de l'équation d'état des gaz parfaits, cette équation rend compte des transitions de phase liquide-vapeur et de l'existence d'un point critique. Elle permet aussi de modéliser l'existence de retards aux changements d'état. Néanmoins, et particulièrement au voisinage du point critique, il sub- siste des écarts notables entre ses prévisions et l'expérience. I--A L'équation de Van der Waals et les mesures au voisinage du point critique. L'équation de Van der Waals relie la pression P, le volume V et la température Td'un fluide en introduisant deux constantes a et b, positives et caractéristiques de ce fluide ; pour une mole, et en notant R la constante des gaz parfaits : [P+%)(V--b)=RT. [I] Page 1/7 Tournez la page S.V.P. Physique 1 -- Filière PSI - 2002 D 1 -- Etablir l'équation de Van der Waals pour n moles de fluide en utilisant des consi- dérations sur le caractère intensif ou extensif des variables. Dans la suite, on raisonnera sur une mole de fluide. D 2 -- La constante b, nommée covolume, représente l'effet de la répulsion à courte distance entre deux molécules. Dans le cas de molécules de faible atomicité, donner un ordre de grandeur numérique raisonnable pour b en vous appuyant sur un modèle simple. Cl 3 -- La figure ci-contre représente, en unités réduites, l'énergie d'interaction Wentre deux molécules, en fonc- tion de la distance r séparant leurs centres de masse. A . --6 grande distance, W oc r . Commenter cette figure ; sau- riez--vous nommer, voire décrire l'origine physique de l'interaction à grande distance ? à très courte distance ? E] 4 -- Quel effet représente la constante a ? Comment justifier le signe positif de a '? Comment interpréter le fait que le terme correctif de la pres- sion (la référence étant le gaz parfait) soit une fonction décroissante du volume '? Cl 5 --L'isotherme critique est celle qui possède dans le diagramme (P, V) un point critique, point d'inflexion de pente nulle. Déterminer en fonction des constantes a, b et R les coordonnées P., V et T du point critique C. Justifier le fait que Van der Waals ait pu prévoir les coordonnées du point critique de l'hélium (T-- -- 5,2 K, P." ---- 2, 275 >< 105 Pa) en 1873, bien avant que la technique ne permette d'atteindre de telles températures. Aurait-il été légitime d'exiger a priori la relation RK = RTC ? _ Ü 6--Commenter le diagramme ci- contre, représentant trois isothermes du fluide décrit par l'équation [l] pour les températures 71 > TC, 73 = T. et 73 < TC. La courbe en pointillés est le lieu des points où (BP) -- '"O. BV Cl 7 -- Représenter dans un diagramme analogue l'allure des isothermes pour un fluide réel quand il ne se produit aucun retard au changement d'état. Faire figurer sur ce dernier diagramme la courbe de saturation. P V [:| 8 ---- Quel est le facteur de compressibilité critiquez C-- "" RT prévu par l'équation de Van der Waals '? On mesure en fait des coefficients z, compris entre 0,29 et 0,31. . ' , . P V T , . Cl 9 -- Les var1ables redu1tes pr = P-- , vr = --- et 9, = ? permettent la representat1on C C C de tous les << fluides de Van der Waals >> par une équation unique, qui s'écrit : oc6 }! ,. Déterminer les valeurs de a, B et y (on trouvera que ce sont des nombres entiers). Page 2/7 Physique 1 ---- Filière PSI - 2002 E] 10 -- Soient p la masse volumique du fluide et pc sa valeur au point critique. Intro- p--a. P--R t * : p. e p P. ligue 5, défini par la propriété suivante, que l'on établira : duisant les écarts réduits p>i< : , trouver la valeur de l'exposant cri-- . . 5 , sur l'isotherme cr1t1que, pour lp*l << 1, p* z D| p*| , ou D est une constante. Cl 11 -- La valeur expérimentale de 5 est ôexp = 4,80i 0,02 ; en quoi la différence p -- 5l ren seigne-t-elle sur le modèle de Van der Waals '? 5 CX [:| 12 -- Proposer un dispositif expérimental permettant de faire passer un échantillon de fluide par un état trèsvoisin du point critique. Quel phénomène optique permet de repérer le passage par le point critique '? Quelle en est l'origine ? I-B Énergie interne, capacité calorifique molaire à volume constant et entropie d'un gaz de Van der Waals. Dans cette section, nous nous proposons de comparer un gaz de Van der Waals au gaz parfait associé. L'énergie interne est notée U et l'entropie S. E] 13 -- À partir de l'expression différentielle du deuxième principe de la thermodyna-- mique, établir l'équation de Helmholtz ci-dessous pour un fluide divariant : ra ------------- 5? ar V Cl 14 ---- En déduire, sous forme d'une intégrale portant sur le volume, la différence entre l'énergie interne d'une mole de fluide et l'énergie interne d'une mole de gaz parfait pris tous deux à la température T et dans le volume V. Cl 15 -- Comment s'exprime cette différence dans le cas du gaz de Van der Waals ? Cl 16 -- Quel est le comportement d'un gaz de Van der Waals dans une détente de Joule-- Gay-Lussac ? Cl 17 -- Comparer les capacités calorifiques molaires à volume constant des deux gaz. El 18 -- En considérant la différentielle de l'énergie libre F = U -- TS , montrer que la différence des énergies libres molaires du gaz de Van der Waals (VdW) et du gaz parfait (GP) s'écrit F@MÏJÜ--ÆJÏJÜ=a%--RTm[V;5} Exprimer, dans la représentation (T,V) , l'équation d'une adiabatique réversible pour une mole de gaz de Van der Waals. I--C Isothermes pour T < T 6, états instables, états métastables et courbe spinodale. On se propose d'établir le théorème de Maxwell qui permet de remplacer l'isotherme déduite de l'équation de Van der Waals par une isotherme présentant un palier de changement d'état. ll s'agit d'abord de trouver la pression à laquelle est observé le changement d'état à la température T, avec T < Tc. On discutera ensuite de la signification des portions de l'isotherme initiale qui ont disparu. Page 3/7 Tournez la page S.V.P. Physique 1 -- Filière PSI -- 2002 Cl 19 -- En se référant au diagramme ci-dessous, donner sans démonstration la relation liant les potentiels chimiques molaires (notés #) des deux phases : liquide au point A et vapeur au point B. Établir la relation entre les aires hachurée de ce diagramme (ce résultat constitue le théorème de Maxwell). On pourra, par exemple, partir de l'expression de la dif-- férentielle du potentiel chimique molaire dans la représentation (T,P) pour des grandeurs molaires : du = ----Sd T + vd P , ou encore exprimer directement L P dv Egg; Il WJÆÏ'ÎÆ. . lez/j ..».- Ifi "" l'...-IH I-------? O. 75 . 1.75 . 0.85 0.8 0.75 0.7 0.65 0.6 Cl 20 -- On admet que les états décrits par l'équation de Van der Waals et correspondant à une valeur négative du coefficient de compressibilité isotherme sont des états thermody-- namiquement instables. Où ces états sont-ils représentés sur l'isotherme dessinée plus haut '? Cl 21 -- Quelle est l'équation P= g(V) de la courbe dite spinodale, située sous la courbe de saturation, qui sépare les états instables des autres états '? '; Cl 22 -- Pour l'eau a = 0,545 m'ÏPa.mol 72 et b = 3,05><10_5 m' .mol_l. Jusqu'à quelle pression maximale, exprimée en atmosphères, peut--on espérer comprimer de l'eau vapeur à T = 1000 C sans observer la fom1ation de gouttes liquides (on prendra R = 8,31 J.K--') '? :::--E E] 23 -- Sous pression assez -V=_' - faible, loin des conditions de , _ liquéfaction et dans un domaine 'A. limité de température, on peut donner prv,«=flvr )pour T= 1,1 Tc àla relation P = f( V) d'une mole de gaz réel la forme, dite du Viriel : Equation du Viriel -- Gaz deVan der Waals ... PV: RT , ABT( ) +.... .. _ - +-- + Gaz parfait V VJ 0 2 4 6 8 10 12 Indiquer, sans développer le calcul, sous quelle condition la relation approchée 86, l 27 1 l _ p,,v, = l+-- 1------ --+ 2 est valide pour le gaz de Van der Waals. 3 3 89 91) , Commenter cette relation (sens, ordres de grandeur...). Fin de cette partie Page 4/7 Physique 1 -- Filière PSI -- 2002 II - Étude de la transition ordre-désordre dans un alliage. Cette partie de l'épreuve étudie quelques aspects thermodynamiques d'un phénomène décrit en 1934 par Bragg et Williams et observé en particulier sur le laiton, alliage de cuivre et de zinc. Au-dessous d'une température 7: nommée température criti- que, les atomes de cuivre et de zinc ne sont plus distribués au hasard sur les noeuds du réseau cristallin, mais se séparent progressivement, sur deux réseaux qui s'interpénètrent. Le cristal est alors dit ordonné. Pour rendre compte de ce phénomène, on traite le problème simplifié d'un alliage binaire constitué de 50 % d'atomes de type A et de 50 % d'atomes de type B, et doté des propriétés suivantes : . Le réseau cristallin com porte un nombre extrê-- mement élevé de sites (et que l'on pourra sans peine supposer pair). Ces N sites sont répartis en deux types notés respectivement sites OL et sites B. Les N/2 sites OL sont les sommets d'un réseau cubique simple. À basse température, ils sont occupés par les N/2 atomes de type A. Les N/2 sites de type B, situés aux centres des cubes définis par les sites de type 0t, forment eux aussi un réseau cubique simple. À basse température, ces sites sont occupés par les N/2 atomes de type B. 0 On néglige toute interaction entre un atome donné et les atomes qui ne sont pas ses plus proches voisins. On néglige également tout effet de bord dans le cristal (N est très grand, de l'ordre de 1024). - L'énergie d'interaction entre deux atomes voisins de natures distinctes est notée VAB. Pour deux atomes de même nature, on la note V,... ou VBB. On néglige toute contribution à l'énergie liée au mouvement des particules. On note pa la probabilité qu'un site 0t donné soit occupé par un atome de type A et ;)B la pro- babilité qu'un site B donné soit occupé par un atome de type B. Dans un cristal désordonné, ..._ _1 pa -- ---- --2--, ce qui signifie que l'occupation des sites est aléatoire. Dans un cristal () () . , 1 ] ordonné, p£' : p£' : 1. Dans un cristal partiellement ordonné, 5 < pa < 1 et 5 < {25 < 1. /7 --z7'"' La relation P= --Qî)ï-- = 2/70, -- 1 définit le paramètre d'ordre, P. Du milieu totalement /7à ' Page 5/7 Tournez la page S.V.P. Physique 1 -- Filière PSI - 2002 désordonné au milieu totalement ordonné, P croît évidemment de 0 à l : 0 S P S 1. CI 23 -- Exprimer en fonction du paramètre d'ordre P et du nombre total d'atomes N : o le nombre NZ d'atomes A sur un site oc, . le nombre N,f d'atomes B sur un site B, o le nombre NÎ d'atomes A sur un site B, o le nombre Ng d'atomes B sur un site oc. Cl 24 -- Exprimer les nombres NAA, NB}, et N,... de liaisons AA, BB et AB obtenues dans le cristal en fonction du paramètre d'ordre P et de N. Pour ce calcul, on remarquera qu'une liaison AA, par exemple, nécessite un atome A en site oc et un atome A dans l'un des 8 sites [3 voisins. Vérifier la relation NAA + NBB + NAB = nombre total de liaisons dans le cristal. Cl 25 -- On pose % = VM + VBB + 2VAB et u1 = VAA + VBB -- 2VAB . Montrer que l'éner-- gie interne du cristal, notée U, peut s'écrire U = Nuo-- NP2 u]. Quelle inégalité est satisfaite entre VAA, VBB et VAE, sachant que les atomes A occupent tous les sites ce au zéro absolu '? En contact avec un thermostat qui lui impose la température T, le cristal subit une évo- lution à volume constant, au cours de laquelle les atomes se redistribuent sur les sites. Dans ce qui suit, on étudie cette redistribution. Cl 26 ---- Sachant que les atomes de même nature sont indiscernables et donc que toute permutation entre eux ne change rien, exprimer, en fonction de P et de N, le nombre de façons distinctes de placer N ? atomes A sur les N/2 sites oc ; exprimer de la même manière le nombre de façons distinctes de placer N 35 atomes B sur les N/2 sites B. Cl 27 -- Calculer le nombre de façons distinctes Q de placer un ensemble de N j atomes A et N £ atomes B sur les N/2 sites B. On devrait trouver Æ . 2 . __ l%l {(1+P)%} {(l--P)%Ï-]l {(l--P)%]l {(1+P)%]f Cl 28 -- En déduire l'expression de l'entropie statistique du cristal S = k ln (Q) , où k est la constante de Boltzmann. Simplifier cette expression en appliquant la formule de Stirling à toutes les factorielles ; formule de Stirling : ln( M!) = M ln( M) -- M . E] 29 -- Admettant l'identité entre l'entropie statistique et l'entropie thermodynamique, établir l'équation vérifiée par le paramètre d'ordre P à l'équilibre thermodynamique. Cet état . . . . . . 2u rend minimale l'énergie libre F = U -- TS . On mtrodu1ra la température critique TL = T' , , . T et la temperature réduite 9 = -7-: . C Page 6/7 Physique 1 -- Filière PSI - 2002 Cl 30 ---- Pour cette question et les suivantes, on pourra éventuellement utiliser la courbe ci-contre. __.-- Montrer que, au voisinage de la température criti-- que, P< TC 1----x 3 l'équation en P établie à la question 28 n'a comme solution que P = O. T Cl 31 -- Établir l'expression de P(9) : P(ï] à très basse température. Donner l'allure C deP(9) pour 0 S GS 1. Cl 32 -- On suppose que le volume du cristal ne dépend pas de la température. Par défi-- 3Ul BU ap -- ---- (attention, P est "a"rÜy " ap ar nition de la chaleur spécifique à volume constant, CV =( le paramètre d'ordre et non pas une pression !). Calculer --io @ dT ; exprimer le résultat en fonction de N, k et TC. Cl 33 -- Calculer la variation d'entropie qui accompagne la transition de phase à T = TL. Cette transition de phase est-elle du premier ordre (associée à une chaleur latente non nulle) ou du deuxième ordre (associée à une chaleur latente nulle) ? Fin de cette partie FIN DE L'ÉPREUVE Page 7/7