
X/ENS Chimie PC 2023
| Thème de l'épreuve | L'état de transition |
| Principaux outils utilisés | cristallographie, cinétique chimique, oxydoréduction, chimie organique, orbitales moléculaires, chimie de coordination, informatique pour tous |
| Mots clefs | état de transition, Eyring, mécanisme, échange de ligands, complexe de nickel, PNP, purines nucléosides phosphorylases, batterie au lithium, GITT |
Corrigé
:👈 gratuite pour tous les corrigés si tu crées un compte
👈 gratuite pour tous les corrigés si tu crées un compte
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
👈 gratuite pour ce corrigé si tu crées un compte
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
Énoncé complet
(télécharger le PDF)













Rapport du jury
(télécharger le PDF)


Énoncé obtenu par reconnaissance optique des caractères
ÉCOLE POLYTECHNIQUE - ESPCI
ÉCOLES NORMALES SUPÉRIEURES
CONCOURS D'ADMISSION 2023
MARDI 18 AVRIL 2023
14h00 - 18h00
FILIÈRE PC - Épreuve n° 4
CHIMIE A (XEULS)
Durée : 4 heures
L'utilisation des calculatrices n'est pas
autorisée pour cette épreuve
L'état de transition est un concept clé en chimie. En effet, une large partie
de la réactivité peut être
interprétée et comprise à partir de sa structure. Cependant, il est extrêmement
difficile d'y accéder, aussi
bien expérimentalement que via des méthodes de modélisation.
Les deux premières parties du sujet illustrent différentes méthodes d'étude de
l'état de transition.
La première illustre la manière dont il est possible d'accéder à des aspects
mécanistiques à l'échelle mi-
croscopique à partir de grandeurs mesurables à l'échelle macroscopique. La
deuxième montre comment
les méthodes de chimie théorique permettent d'accéder à la structure associée à
l'état de transition pour
ensuite mieux comprendre les différents mécanismes possibles.
Ensuite, le sujet s'attache à montrer en quoi la connaissance de la structure
de l'état de transition
permet de concevoir de nouveaux matériaux ou molécules : la troisième partie
détaille ainsi comment il
est possible de synthétiser un inhibiteur enzymatique qui vient bloquer le site
actif d'une protéine, tandis
que la dernière partie montre comment la caractérisation de l'état de
transition permet d'envisager des
batteries innovantes qui puissent s'affranchir du lithium.
Chaque partie est indépendante et peut être traitée séparément. Des données
numériques ainsi
que des documents annexes (tables spectroscopiques, banque de réactions) sont
regroupés à la fin du
sujet.
1 Détermination du mécanisme réactionnel pour une réaction d'échange de
ligand
Le but de cette partie est de déterminer le mécanisme réactionnel prépondérant
pour un échange
de ligand pour des complexes hexaaqua du groupe 13 de la classification
périodique : [AI(H2O})6]°*,
[Ga(H20)}6/* et [In(H20)6]**. La réaction générale correspondante est
représentée figure 1 :
-- --19+
OH C OH 7
| Oh | OF
H}20--MS--OH: | + HO > | H}0--M----*OH: | + HO
HO | l (1) HO | L (1)
OH; OH;
-- -- (ad) L _{(aq)
FIGURE 1 -- Représentation de la liaison {1 rompue et de la liaison {2 formée
lors de la réaction
d'échange de ligand autour de l'ion central M = (Al; Ga; In). L'exposant * sert
uniquement à distinguer
la molécule d'eau échangée lors de la réaction.
La réaction d'échange de ligand correspond à la rupture d'une liaison { « ion M
»/eau et à la
création d'une nouvelle liaison {, « ion M »/eau*. Ce type de réaction est
particulièrement difficile à
étudier car le ligand échangé est également le solvant.
Un des outils qui permet de faire le lien entre grandeurs macroscopiques et
mécanistiques est la
formule d'Eyring (ou théorie de l'état de transition). Cette théorie a permis
d'aboutir à une formulation
de la constante de vitesse k pour un acte élémentaire, se déroulant à
température T et pression P fixées :
__ kBT AG} An
k-- exp (+) x (c°) (1)
où Kg est la constante de Boltzmann, h la constante de Planck, KR la constante
des gaz parfaits, AG?
l'enthalpie libre d'activation molaire, c° la concentration standard et An
l'opposé de la somme des coef-
ficients stæchiométriques algébriques -- ce terme permet d'avoir une constante
de vitesse ayant la bonne
dimension en fonction de l'ordre de la réaction.
-- 1/23 -
© OO I OO OO & À NN H
NO NN NN NN NN NNR RER BB PB BB HA
© © -- OO O0 © NN A OO GO OO --J OO O0 À NN) A O
30
On admettra que toutes les relations thermodynamiques vues pour l'enthalpie
libre restent valables pour
l'enthalpie libre d'activation. Dans ce sujet on se place toujours dans
l'approximation d'Ellingham : l'enthalpie
d'activation AH et l'entropie d'activation ASY sont indépendantes de la
température.
1. Donner l'expression de la constante de vitesse donnée par la loi d'Arrhénius
et préciser les grandeurs
introduites. Indiquer l'intérêt de la formule d'Evyring par rapport à la
formulation que vous venez
de donner.
2. Montrer qu'une régression linéaire portant sur la grandeur ET permet
d'extraire l'entropie et l'en-
B
thalpie d'activation à partir des valeurs de k à différentes températures.
Le code donné figure 2 est une ébauche de script Python permettant de faire la
démarche proposée
à la question 2 pour l'ion aluminium(IIl). Une fois complété et exécuté, le
script affiche le graphique
correspondant à la figure 5.
"1 Script pour obtenir les grandeurs d'activation pour l'ion
[A1(H_20)_{6}]7{3+} """
MM Importation des librairies """
import numpy as np
import matplotlib as mpl
import matplotlib.pyplot as pit
import scipy.constants as constants
k_B = constants.k # constante de Boltzmann en Unités du Système International
(USI)
R = constants.R # constante des gaz parfaits en USI
h = constants.h # constante de Planck en USI
"1 Températures en Kelvin """
T = np.asarray([394.5,391.2,386.9,377.7,371.3,367.8,362.9])
x = 1/T
"1 constantes de vitesse mesurées expérimentalement en s°-1 """
k = np.asarray([5750,4582,3199,1832,1148,816,553])
y =
"M" régression linéaire """
a, b =
y_fit = a*x+b
"M" Affichage des valeurs finales """
DeltaHactivation =
DeltaSactivation =
"1 tracé des points bruts """
plt.scatter(x,y,color="black" ,marker='+)
"M tracé de la régression linéaire """
plt.plot(x,y_fit, label='$\Delta,
H\ddagger={}"\mathrm{{kJ\cdot;:mol®{{-1}}.}}";7\Delta, ;S7\
ddagger={}"\mathrm{{J\cdot, K°{{-1}},\cdot,
mol"{{-1}}}}$'.format(DeltaHactivation,
DeltaSactivation) ,color="black")
plt.show()
FIGURE 2 -- Ébauche de script Python pour extraire l'enthalpie d'activation et
l'entropie d'activation à
partir de données expérimentales pour l'échange d'une molécule d'eau pour le
complexe
hexaaquaaluminium(IIT) [AI(H20)6[°*.
3. Compléter la ligne 15 pour calculer la grandeur y sur laquelle va porter la
régression linéaire. Com-
pléter ensuite la ligne 18 pour effectuer la régression linéaire. On pourra
s'aider de la documentation de
certaines fonctions fournie en page 20.
-- 2/23 --
-- AHi= 837 kJ-mol !; ASf=37].K-!.mol !
--21,5 -
--22,0 -
--22,5 -
--23,0 -
F
0,002650 0,002700 0,002750
1/T
0,002550 0,002600
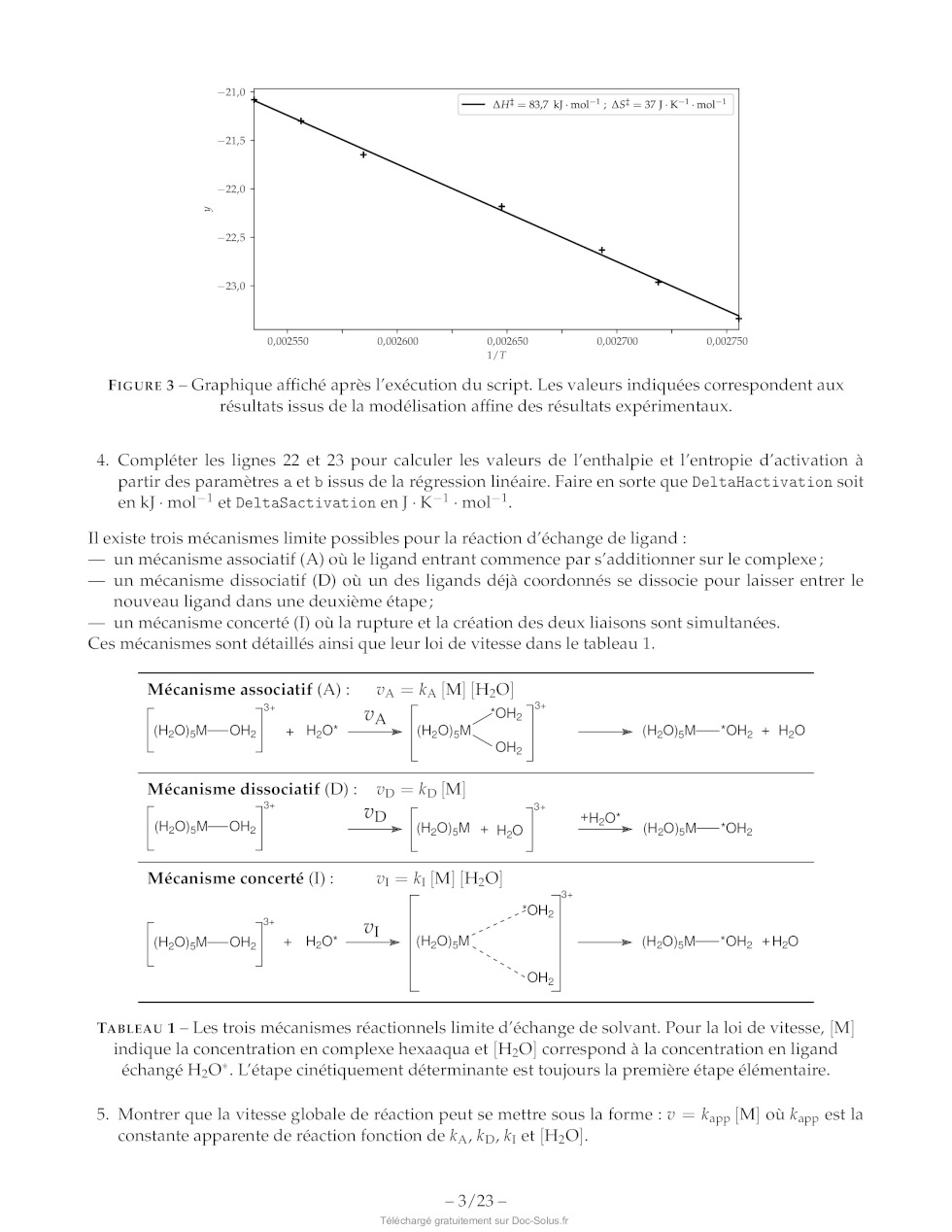
FIGURE 3 -- Graphique affiché après l'exécution du script. Les valeurs
indiquées correspondent aux
résultats issus de la modélisation affine des résultats expérimentaux.
4. Compléter les lignes 22 et 23 pour calculer les valeurs de l'enthalpie et
l'entropie d'activation à
partir des paramètres a et b issus de la régression linéaire. Faire en sorte
que DeltaHactivation soit
en kJ :mol let DeltaSactivationen]:K-!:mol !.
Il existe trois mécanismes limite possibles pour la réaction d'échange de
ligand :
-- un mécanisme associatif (A) où le ligand entrant commence par s'additionner
sur le complexe;
-- un mécanisme dissociatif (D) où un des ligands déjà coordonnés se dissocie
pour laisser entrer le
nouveau ligand dans une deuxième étape;
-- un mécanisme concerté (I) où la rupture et la création des deux liaisons
sont simultanées.
Ces mécanismes sont détaillés ainsi que leur loi de vitesse dans le tableau 1.
Mécanisme associatif (A): va -- ka [M] [HO]
3+ UA _ OH 3+
(H50)5M-- OH, + H50O* ----> (H2O)SM --> (H50)5M----*OH; + H50
OH
Mécanisme dissociatif (D): vp = kp M]
3+ 3+
UD +H20*
HLON--0n ----> CS + 40 2% (H)0)};sM--*OHs
Mécanisme concerté (l) : 0 = k [M] [HO]
-- --3+
- OH
3+ VI Let 0 2
{HOMO + HO ------ (H20)5M ----+ (H50)5M--*OH; +H,0
OH
TABLEAU 1 -- Les trois mécanismes réactionnels limite d'échange de solvant.
Pour la loi de vitesse, [M]
indique la concentration en complexe hexaaqua et [H20! correspond à la
concentration en ligand
échangé H20*. L'étape cinétiquement déterminante est toujours la première étape
élémentaire.
5. Montrer que la vitesse globale de réaction peut se mettre sous la forme : v
= k;pp [M] où kapp est la
constante apparente de réaction fonction de KA, kp, kr et [H20|.
-- 3/23 --
En pratique, la constante apparente K;,, est mesurée par spectroscopie RMN. Il
est ensuite possible
d'identifier le mécanisme prépondérant à partir de la valeur de l'entropie
d'activation.
6. Pour chacun des mécanismes limite, prédire si l'entropie d'activation
attendue est x) fortement né-
gative, 6) fortement positive, ou y) faible en valeur absolue.
On peut également calculer un volume d'activation AV. Il correspond à la
variation de volume entre
les réactifs et l'état de transition. Les grandeurs d'activation étant en tout
point analogues aux grandeurs
classiques, l'expression de la différentielle de l'enthalpie libre d'activation
vérifie la relation :
d AG? = AVfdP -- ASÏdT (2)
7. En déduire qu'en mesurant la constante de vitesse à différentes pressions,
il est possible d'accéder au
volume d'activation et donner l'expression AV en fonction de la constante de
vitesse k, de la pres-
sion P, de la température T et de la constante des gaz parfaits KR. On
supposera le volume d'activation
indépendant de la pression.
Dans le cadre de l'étude, le volume d'activation a le même signe que celui
attendu pour l'entropie d'ac-
tivation (question 6). De plus, il est faible ou nul quand l'entropie
d'activation l'est également.
Il est possible de représenter le mécanisme réactionnel dans un diagramme à
deux dimensions appelé
diagramme de More-O'Ferrall pour indiquer le degré de rupture de la liaison {,
et de création de la
liaison {> dans l'état de transition (les liaisons {1 et {2 sont représentées
sur la figure 1). Pour le tracé,
on suppose que la nouvelle liaison créée {2 est d'autant plus forte que la
liaison {1 est rompue. Cela
correspond au fait de placer obligatoirement l'état de transition sur la
diagonale de la représentation à
deux dimensions. C'est le volume d'activation mesuré qui permet alors de placer
l'état de transition sur
la diagonale.
Un diagramme de More-O'Ferrall pour chacun des trois mécanismes limite
présentés tableau 1 est donné
figure 4 a). Le diagramme réel pour les trois ions étudiés est donné figure 4
b).
À) CCR, b) x CR:
Réactifs-'
Y In°t
État de
$
,. ee ©
transition .*#-------------.. Y
Y a
15
3+
1CA
\? > > > N" )
AV? CR AV? CR,
Produits
FIGURE 4 -- a) Diagramme de More-O'Ferrall pour les trois mécanismes limite
présentés tableau 1. b)
Diagramme pour les trois ions AT, Ga°* et In°**. Le volume d'activation est
donné sur la diagonale en
mL - mol-! ; les nombres 13,5; 0; --13,5 correspondent à la valeur de AVY aux
points A,BetC
respectivement.
8. Déduire à quels mécanismes limite (voir tableau 1) correspondent
respectivement les chemins pas-
sant par les points À, B et C -- sur la figure 4 a). En déduire si la
coordonnée de réaction CR; corres-
pond à la création de la liaison {> ou à la rupture de la liaison #1.
-- 4/23 --
9. Pour le mécanisme étudié, justifier à l'aide d'un calcul simple que les
valeurs extrêmes du volume
-- 13,5 mL:mol {.
d'activation sont de l'ordre de grandeur de ceux indiqués figure 4 : | AVEax
10. À l'aide de la figure 4 et de la question 8, déduire le type de mécanisme
privilégié pour chacun des
ions A+, Ga°+ et In°+.
11. Pour l'ion Al°*, indiquer si la valeur expérimentale de l'entropie
d'activation : 37 J - K_! : mol {est
en accord avec les prédictions faites à la question 6.
12. En vous aidant des rayons ioniques fournis dans les données, proposer une
interprétation microsco-
pique qui justifie la différence de mécanisme limite observé pour ces trois
ions appartenant tous à la
même colonne du tableau périodique.
2 Intermédiaire réactionnel et compréhension de la nature des interactions
orbitalaires
En pratique, il est extrêmement difficile de caractériser expérimentalement le
complexe activé qui corres-
pond à la structure géométrique de l'état de transition. Les simulations
numériques sont maintenant des
méthodes de choix pour caractériser microscopiquement l'état de transition.
C'est ensuite la cohérence
avec les données expérimentales qui permet d'entretenir un dialogue
théorie-expérience pour aboutir à
une compréhension détaillée des systèmes.
On s'intéresse dans cette partie à un complexe de nickel
[Ni(H2)(B-Ph)(P-(iPr)2Ph)2|, noté 1, capable de
rompre la liaison du dihydrogène. L'utilisation du nickel est intéressante car
c'est une très bonne alterna-
tive au palladium ou au platine sur le plan économique. Des études récentes ont
permis de montrer que,
pour cette famille de complexes, le ligand joue un rôle important pour couper
la liaison du dihydrogène.
b) vue « de profil »
a) vue « de face »
FIGURE 5 -- Deux vues du complexe de nickel étudié, noté 1 : a) de face, b) de
profil. Pour la
représentation tridimensionnelle, tous les atomes d'hydrogène autres que ceux
directement liés à
l'atome de nickel sont omis pour plus de clarté. Pour le complexe modèle, seule
la première sphère de
coordination est représentée.
Dans le complexe 1, outre le dihydrogène, l'atome de nickel est lié à deux
atomes de phosphore (notés
P: et P2) et à un atome de bore (figure 5).
On modélise le complexe réel par un complexe simplifié (figure 5) ayant un plan
de symétrie qui est le
plan xOz. Dans le complexe modèle, l'atome de nickel est au centre du repère,
l'atome de bore est dans
le plan xOz et la molécule de dihydrogène est dans le plan yOz. La méthode des
fragments sera utilisée
pour construire les orbitales moléculaires du complexe modèle.
Une fois ce modèle construit, il sera possible de le confronter au résultat
d'un calcul mené avec des
méthodes de chimie théorique poussées pour comprendre les interactions ayant un
rôle déterminant au
sein du complexe.
-- 5/23 -
13. Donner le diagramme d'orbitales moléculaires de H} par interaction entre
les orbitales de valence
des atomes d'hydrogène. L'orbitale la plus basse en énergie sera notée ÿ et la
plus haute en énergie
sera notée 2. Indiquer le remplissage électronique et représenter les orbitales
moléculaires 1 et d2
formées.
14. Indiquer si les orbitales 1 et #2 du dihydrogène sont symétriques ou
antisymétriques par rapport
au plan xOz.
Pour l'atome de nickel, seules les orbitales d sont considérées. Elles sont
représentées figure 6 a).
d 2-1 d 22
a)
b)
FIGURE 6 -- a) Surface d'isodensité pour les orbitales d de l'atome de nickel.
b) Représentation des trois
orbitales du fragment « BP;:P; » prises en compte pour étudier le complexe
modèle. La représentation
est une vue « de profil » comme sur la figure 5 b).
15. Pour chacune des orbitales d de l'atome de nickel, indiquer si elle est
symétrique ou antisymétrique
par rapport au plan xOz.
Un autre fragment est constitué avec les atomes de la première sphère de
coordination : « BP;:P; » dont
on se limite à quelques orbitales. Les ligands phosphorés sont des ligands
nucléophiles, l'interaction
avec le nickel se fait donc essentiellement via des orbitales occupées
(orbitales #3 et #4, figure 6 b)). Pour
sa part, le bore intervient via une orbitale vacante (5, figure 6 b)).
16. Pour chacune des orbitales ÿ3 à ds, indiquer si elles sont symétriques ou
antisymétriques par rapport
au plan xOz.
17. Indiquer tous les recouvrements possibles entre les orbitales #; et chacune
des orbitales d. Pour cela,
présenter les résultats sous la forme d'un tableau analogue au tableau 2,
mettre un zéro lorsque le recouvrement
est nul par symétrie et un symbole V sinon.
ÿ
Y5
TABLEAU 2 -- Forme attendue pour le tableau-réponse de la question 17.
18. Pour chacune des orbitales du dihydrogène (ÿ1 et 2), caractériser le type
de recouvrement possible
avec le métal (axial ou latéral). Pour chaque interaction, indiquer si le
dihydrogène se comporte
comme un nucléophile ou un électrophile.
-- 6/23 --
FIGURE 7 -- Orbitales de valence pour le complexe 1 (de la HO--4 à la BV). Tous
les atomes d'hydrogène
autres que ceux directement liés à l'atome de nickel sont omis pour plus de
clarté.
Sans calcul, il est impossible de quantifier précisément quelles sont les
interactions prépondérantes
parmi celles identifiées précédemment. Le diagramme d'orbitales moléculaires
obtenu avec des mé-
thodes de calcul avancées est donné figure 7. Il permet d'avoir une vision plus
précise de la force relative
des interactions.
19. Pour les orbitales Y; et Y3 uniquement, identifier l'interaction
prépondérante parmi celles proposées
à la question 17. Indiquer pour chaque orbitale si l'interaction entre l'atome
de nickel et le dihydro-
sène est liante, non-liante ou antiliante.
20. À l'aide de la figure 7, expliquer pourquoi la liaison H,-H4 passe de 0,740
À pour le dihydrogène
libre à 0,837 A lors de la formation du complexe 1.
21. Donner la configuration électronique d'un atome de nickel dans son état
fondamental. Sachant que
tous les électrons de la figure 7 peuvent être considérés comme des électrons
de valence du nickel,
en déduire son degré d'oxydation -- rare pour la géométrie observée.
-- 7/23 -
Le complexe 1 peut ensuite évoluer pour aller jusqu'à la rupture de la liaison
H-H pour former le com-
plexe 2 (figure 8). Les auteurs de l'étude ont ainsi cherché la structure de
l'état de transition pour mieux
comprendre les interactions orbitalaires mises en jeu pour rompre cette
liaison. Ils ont pu identifier deux
mécanismes concurrents pour passer du complexe 1 au complexe 2:
-- Le mécanisme 1 passe par un unique état de transition ET1 : 1--ET1--2;
-- Le mécanisme 2 passe par un premier état de transition appelé ET2' puis un
intermédiaire réactionnel
Int puis passe par un deuxième état de transition ET2 : 1--ET2'--Int-- ET2--2.
AT Mécanisme 1
CR
Mécanisme 2 Pr PQ
iPr P
_/ SEP , /
A RE) DA
ET2' ET2
FIGURE 8 -- Structure schématique des complexes 1 et 2 avec la succession
d'entités pour passer de l'un à
l'autre lors des deux mécanismes possibles.
iPr
Mécanisme 1 Mécanisme 2
Distance (À) 1 ET1 2 1 ET2 Int ET2 2 dihydrogène
H,-Hp 0,837 1,977 3,079 0,837 0,836 0,841 1,017 3,079 0,740
B-Hy 3,730 3,209 1,312 3,730 2,121 2,012 1,643 1,312
Ni-Hy, 1,660 1,440 1,619 1,660 1,661 1,663 1,588 1,619
TABLEAU 3 -- Distances sélectionnées pour les différentes structures. Toutes
les distances sont en
Angstrôm. La longueur de liaison pour le dihydrogène libre est également donnée.
22. À l'aide du tableau 3, décrire l'évolution des distances H,-H, et Ni-H, et
préciser l'étape correspon-
dant à la rupture de la liaison H,-H, pour chacun des deux mécanismes.
23. Décrire chacun des deux chemins réactionnels en précisant autant que
possible les rotations, liai-
sons formées et rompues pour passer d'une structure à l'autre. On pourra entre
autres s'aider de
l'évolution de la distance B-H..
24. À partir du tableau 3 et des différentes données, nommer la catégorie de
réaction qui correspond
au passage de 1 à 2 pour le mécanisme 1 -- parmi les grandes familles de
réactions couramment
rencontrées dans les cycles catalytiques.
3 Contrôle de la cinétique des purines nucléosides phosphorylases
Les purines nucléosides phosphorylases (PNP) sont des enzymes capables de
convertir des purines
nucléosides, comme l'inosine, en purine correspondante ainsi qu'en du
ribose-1-phosphate (figure 9).
Comme l'activité de ces enzymes intervient dans des cycles cellulaires, ce sont
des cibles importantes
pour le développement de traitements en chimiothérapie ainsi que pour la
synthèse de molécules d'in-
térêt biologique.
-- 8/23 -
J=N Ô 0®
N O O /
EUR NE + 407 7
| OH
7 N AZ LS O
HO OH H HO OH
inosine hypoxanthine ribose-1-phosphate
I
O
À
Z
+
O
T
ND
©
O
N< FIGURE 9 -- Exemple de réaction catalysée par les PNP sur l'inosine. 3.1. Structure de l'enzyme et interactions avec le substrat Pour pouvoir contrôler l'activité enzymatique, il est nécessaire de connaître la structure du complexe enzyme-substrat correspondant à l'état de transition. Cette structure est présentée figure 10 a). Son ana- lyse permet ensuite de concevoir des molécules ayant une structure adaptée au site enzymatique qui puissent jouer le rôle d'inhibiteurs. HN 257 NH \ n N dé | O 243 $o H N O OH O 201 O H H N 50 VA O FIGURE 10 -- a) Structure tridimensionnelle de l'inosine dans la poche enzymatique. Seules les chaînes latérales des acides aminés au contact de l'inosine sont représentées -- les atomes d'hydrogène sont omis. b) Structure schématique correspondante -- l'état de protonation n'étant pas représentatif de la structure réelle à pH = 7. Les numéros sur la partie enzymatique indiquent la position de l'acide aminé le long de la structure primaire de la protéine et l'inosine est en bleu. 25. Identifier les acides aminés aux positions 86, 200 et 201 le long de la chaîne primaire --- représentés sur la figure 10 b). 26. Décrire l'état de protonation majoritaire à pH = 7 de chacune des chaînes latérales des acides aminés aux positions 86, 200 et 201 représentés figure 10 b). 27. Pour les acides aminés aux positions 86, 200 et 201 représentés figure 10, indiquer les types d'inter- action moléculaire qu'ils sont capables d'établir avec le substrat inosine. 3. II. Synthèse d'un inhibiteur des enzymes de type PNP À partir de l'étude de la structure du complexe enzyme-substrat, l'immuciline H a été identifiée comme inhibiteur potentiel des enzymes de type PNP - figure 11. Sa structure étant proche de celle du substrat dans l'état de transition, elle forme un complexe stable avec la protéine. Cela permet d'inhiber l'action de l'enzyme et d'assurer une régulation qui peut apporter des améliorations sur le plan physiologique. -- 9/23 - =N H NH ON Lo NY O HO À NY HO 7 Hô OH 7 HÔ OH 7 inosine immuciline H FIGURE 11 -- Structure de l'inosine, substrat des enzymes de type PNP, et de l'immuciline H, qui les inhibe. Cette partie s'intéresse à la synthèse de l'immuciline H. La première partie correspond à la synthèse d'un dérivé de l'iminoribitol 1 à partir de la gulono-1,4-lactone 8 (figure 12). n So Bn 5 HOT : 40) e e CT 6. O 6. O 6. O 6. O K 4 K 2 X 3 K 4 VC le 1) NaBH4, EtOH-H,0 (5:1), 0 °C, 30 min So So 2) CH3SO,CI, DMAP (substæch.), pyridine, t.a., 2h A B 3) H>, Pd/C, EtOH, ta., 2h 0). (7° O7. 7° O7. Cr
4) LiAIH4 THF-Et,0 (9:1), t.a., 30 min :
5) PACHNH, 60 °C, 60h HO OH Ô O Ô O
6) 2,2-diméthoxypropane, APTS, acétone, t.a., 2 jours 8 X 7 X 6
ZT
(us
>
ZW
FIGURE 12 -- Schéma rétrosynthétique 1 = 8. Le symbole -- indique que la
molécule à gauche de la
flèche a été synthétisée à partir de la molécule à droite de la flèche. Tous
les acronymes sont explicités à
la fin du sujet.
28. Faire correspondre les étapes (A-F) du schéma rétrosynthétique donné en
figure 12 aux conditions
opératoires 1) à 6) qui y sont détaillées. Aucune justification n'est attendue.
On admettra que les condi-
tions 4) sont utilisées avant les conditions 1) dans l'ordre chronologique de
la synthèse.
29. Proposer d'autres conditions opératoires pour effectuer la transformation 8
-- 7. Donner le méca-
nisme correspondant à cette proposition.
30. Représenter le mécanisme schématique de la transformation 7 -- 6. Pour
cela, le réactif utilisé pourra
être assimilé à un ion hydrure.
31. Donner l'équation de réaction de la transformation 6 -- 5. Commenter le
fait de ne pas opérer en
milieu acide lors de cette réaction.
32. Représenter le mécanisme réactionnel le plus probable de la transformation
5 -- 4 au vu des struc-
tures des réactifs et du produit. Indiquer en quoi les conditions
expérimentales sont cohérentes avec
le mécanisme réactionnel limite représenté.
-- 10/23 -
ü mise en place û NCS N Li
HOT Pto Po }
Le du groupe Le pentane, LE THF, -78 °C,
Le protecteur P' x ta.,1h O, _O 5 min
1 9 10
P? N
| NX -CO:Et
16 CbzCI, DBU, NT
_--. à PO CN
anh. CH,Cb, i 2
À, 4h es
15
?
© ©
AcO NH; p?
J | NH
17 HN NH N. / O
-- PO Z
EtOH, A, 16h NE NH
Ce
18
1) HO: AcOH(10:1)
_ ta., 15h
O ©
2) CI HN COLE
NaOAc, MeOH ,
ta. 16h
N
de
11
du groupe
protecteur P?
P£ NMe; P£
ND ? N
Po CN p! 07EUR } CN
%
14
mise en place |
H NH
? N. / O
HO T
Le NE NA
HÔ OÙ
immuciline H
FIGURE 13 -- Schéma de synthèse 1 -- immuciline H. Les groupes P! et P? sont
des groupements
protecteurs qui sont discutés dans le texte. Tous les acronymes sont explicités
à la fin du sujet.
La synthèse de l'immuciline H peut ensuite se faire à partir de la molécule 1
en 11 étapes (figure 13).
Pour les étapes marquées d'un « ? », les conditions opératoires font l'objet de
questions au sein du sujet.
33. La transformation 9 -- 10 est une réaction d'oxydo-réduction. Équilibrer la
demi-équation électro-
nique correspondant au couple formé par les espèces 9 et 10, puis en identifier
l'oxydant et le réduc-
teur.
34.
Identifier, en justifiant la réponse à l'aide des conditions expérimentales, le
mécanisme réactionnel
limite le plus probable de la f-élimination 10 -- 11, et le représenter.
39.
36.
Donner la structure de deux produits indésirables qui pourraient se former lors
de l'étape 10 -- 11.
Sachant que la fonction imine C=N possède une réactivité analogue à celle d'un
dérivé carbonylé,
prévoir un réactif unique utilisable pour synthétiser l'espèce 12 à partir de
l'imine 11. Proposer un
mécanisme réactionnel pour la transformation 11 -- 12, hors considérations
stéréochimiques.
-- 11/23 -
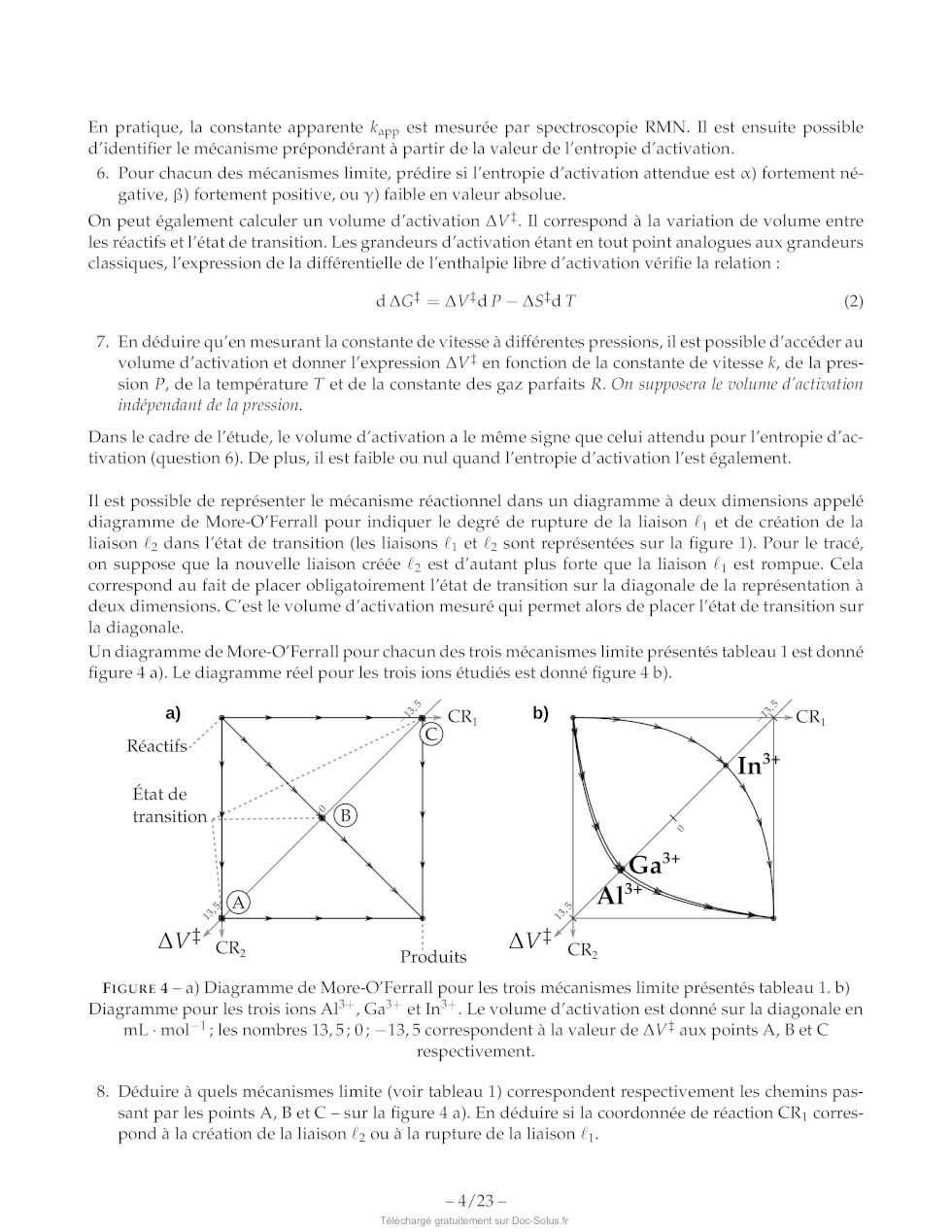
FIGURE 14 -- Vues tridimensionnelles de la molécule 11.
37. Donner une représentation de Newman de l'imine 11 selon la liaisons C3-C4
en plaçant l'atome C3
au premier plan. Il est possible de s'aider de la figure 14.
38. Justifier la stéréochimie du centre stéréogène généré lors de la formation
de l'espèce 12, et fournir
son stéréodescripteur absolu en le justifiant.
Par la suite, on admettra que le groupement nitrile -C=N confère des propriétés
analogues à celles d'un
groupement carbonyle, les propriétés acido-basiques en particulier. De même, le
motif C=C-C=N a une
réactivité similaire à celle d'une x-énone.
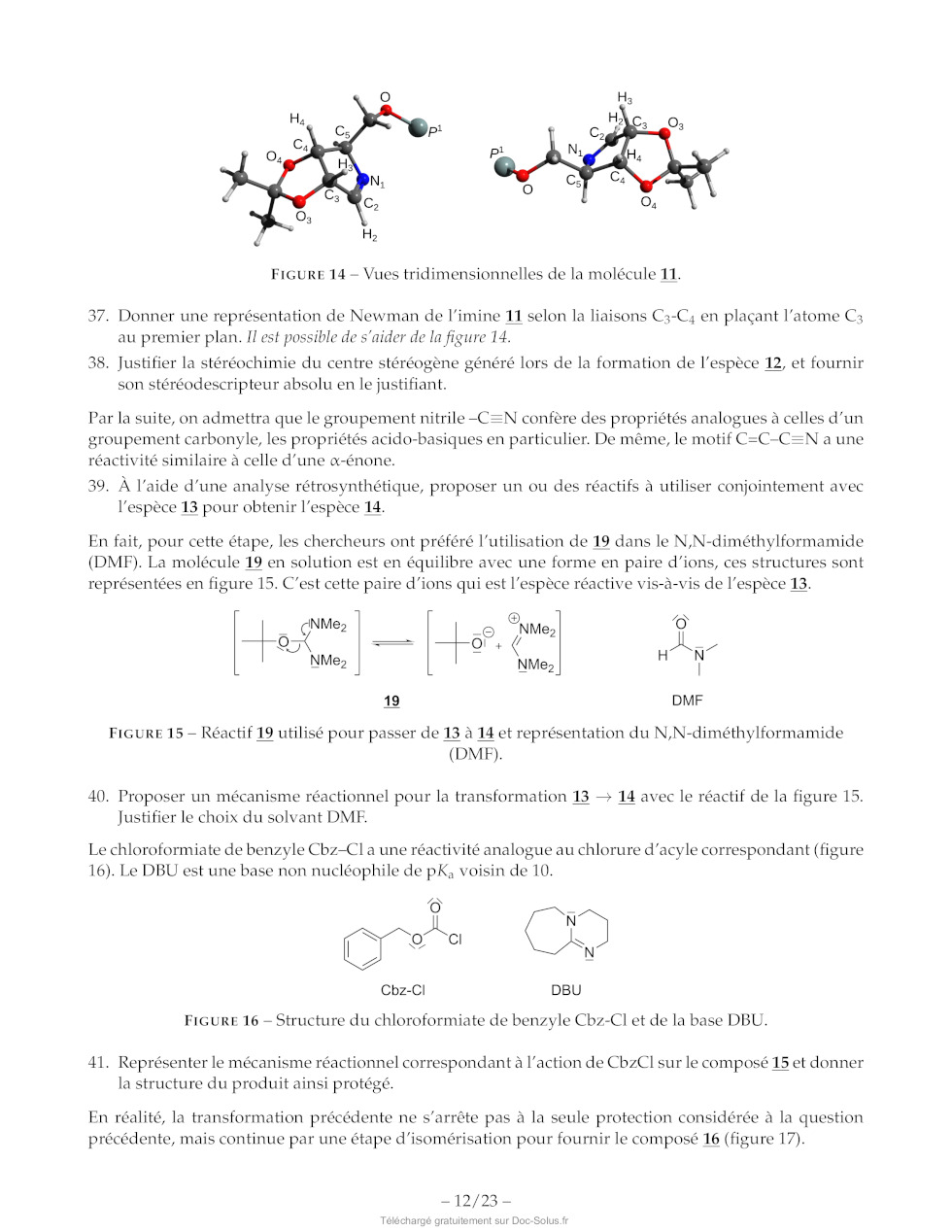
39. À l'aide d'une analyse rétrosynthétique, proposer un ou des réactifs à
utiliser conjointement avec
l'espèce 13 pour obtenir l'espèce 14.
En fait, pour cette étape, les chercheurs ont préféré l'utilisation de 19 dans
le N,N-diméthylformamide
(DMEF). La molécule 19 en solution est en équilibre avec une forme en paire
d'ions, ces structures sont
représentées en figure 15. C'est cette paire d'ions qui est l'espèce réactive
vis-à-vis de l'espèce 13.
@ NT
h çINMe; _© NMe> O
La") [0 À
NMe; NMe> |
19 DMF
FIGURE 15 -- Réactif 19 utilisé pour passer de 13 à 14 et représentation du
N,N-diméthylformamide
(DME).
40. Proposer un mécanisme réactionnel pour la transformation 13 -- 14 avec le
réactif de la figure 15.
Justifier le choix du solvant DMF.
Le chloroformiate de benzyle Cbz-Cl a une réactivité analogue au chlorure
d'acyle correspondant (figure
16). Le DBU est une base non nucléophile de pK, voisin de 10.
9 "a OX
Cbz-Cl DBU
FIGURE 16 -- Structure du chloroformiate de benzyle Cbz-Cl et de la base DBU.
41. Représenter le mécanisme réactionnel correspondant à l'action de CbzCTI sur
le composé 15 et donner
la structure du produit ainsi protégé.
En réalité, la transformation précédente ne s'arrête pas à la seule protection
considérée à la question
précédente, mais continue par une étape d'isomérisation pour fournir le composé
16 (figure 17).
-- 12/23 ---
Cbz
2 /
PT SN
1 N 6 8
2 y KE 2
PO 7 --COOE!
5 2: INK
O 3 O 10
XX
12 11
FIGURE 17 -- Stucture du composé 16.
42. Montrer que les signatures spectroscopiques du composé 16 fournies dans les
tableaux 4 et 5 sont
compatibles avec la structure proposée figure 17. En RMN, se limiter aux 5
signaux -- À, B,E, H et I -
qui ne sont pas des multiplets. Lorsque nécessaire, utiliser la numérotation
des atomes donnée figure 17 pour
indiquer les liaisons concernées (IR) ou les protons liés à l'atome numéroté
(RMN).
Signal A B C D E F G H I
ô (ppm) 7,00 5,54 4,85 4,26 4,19 3,40 3,04 1,42 1,22
Intégration 1H 2H 2H 2H 2H 1H 1H 6H 3H
Multiplicité s slarge m m q m m s t
Constante de couplage -- -- --- --- 71Hz --- --- -- 7,1 HZ
TABLEAU 4 -- Spectre RMN du proton simplifié du composé 16 : les signaux
correspondant aux groupes P!,
P? et Cbz ont été retirés. Abréviations : s singulet, t triplet, q quadruplet,
m multiplet.
(cm!) 3422 3374 3086-2867 1722 1478 1190 1022
Intensité moyenne moyenne moyenne forte moyenne faible faible
TABLEAU 5 -- Spectre infrarouge simplifié du composé 16 : les signaux
correspondant aux groupes P}, P? et
Cbz ont été retirés.
43. La transformation 16 -- 17 correspond à la déprotection du groupement Cbz.
Elle se produit dans
les mêmes conditions que la réaction d'hydrogénation d'un alcène en catalyse
hétérogène, et fournit
du toluène et du dioxyde de carbone comme sous-produits. Donner les conditions
expérimentales
nécessaires pour cette étape de déprotection.
44. Justifier l'intérêt des groupes protecteurs Pl et P? utilisés lors de la
synthèse de l'immuciline H.
45. À l'aide du tableau 9 fourni dans les annexes, proposer deux groupes
protecteurs Pl et P? compa-
tibles avec la synthèse effectuée.
46. Proposer des conditions opératoires successives qui permettent d'obtenir
l'immuciline H à partir de
18. Les réponses devront être justifiées.
Une voie alternative de synthèse de l'immuciline H est donnée figure 18.
L'étape 20 -- 21 y est cruciale
car sa diastéréosélectivité impacte fortement le rendement final. C'est le
diastéréoisomère 21f qui est
le composé souhaité. Pour cela, les chercheurs ont privilégié l'utilisation du
composé H;3BSMe> pour
réaliser cette étape.
47. Donner la structure de Lewis de l'espèce H3BSMez. Donner la géométrie
locale des atomes de bore
et de soufre.
En solvant organique, l'espèce H3BSMe; est en équilibre avec le borane BH.
-- 13/23 --
NH H NH H NH
N, / OMe , tif testé NO. / OMe N / OMe
HO a réactit este, Ho } 77 . HO TT
5 2 N&EAN N 5 2 N&EAN 5 5 NEAN
N 20 À 214 \ 28
O
NS bé O
NS EH & 080 ® B_
réactif 5H CH O LH O J'OBCH
testé | >
osté ONe où T° où 4
NaBH, NaBH3CN L-Sélectride STAB MeCBsS H3BSMe,;
rapport 1:1 1:1 4:1 4:1 4:1 1:2
210:21B
FIGURE 18 -- Transformation 20 -- 21, réactifs testés et diastéréosélectivités
observées.
48. Par analogie avec la réactivité connue du borane BH sur les alcènes,
représenter un intermédiaire
réactionnel qui permette de rendre compte de la réduction 20 -- 21, hors
considération de stéréochi-
mie.
Dans le but d'augmenter la proportion d'isomère 218 les chercheurs ont d'abord
testé un ensemble
d'espèces, qui sont toutes des hydrures de bore à la réactivité similaire au
tétrahydruroborate de sodium
NaBH, (figure 138).
49, Justifier l'évolution de la diastéréosélectivité de la transformation en
fonction du réactif utilisé. En
particulier, identifier un type d'interaction spécifique entre le borane et la
molécule considérée pour
faire l'hydruration par la face la plus encombrée.
4 Importance de la structure et de la morphologie pour des batteries au li-
thium
Le stockage de l'énergie est un enjeu critique pour de nombreuses applications,
en particulier pour les
appareils électroniques comme les ordinateurs ou les smartphones. La plupart
des batteries actuellement
utilisées exploitent le lithium. Dans tous les matériaux utilisés, le lithium
est capable de s'insérer de
manière réversible pour former un accumulateur. Pour mieux comprendre les
propriétés de ces batteries,
il est important de connaître le mécanisme de déplacement des ions dans la
structure et donc d'être
capable d'identifier les états de transition lors de la migration des ions au
sein du matériau.
Parmi les différentes structures présentant un intérêt, l'oxyde mixte de titane
et de niobium TiNb207 est
très prometteur. Il peut accepter jusqu'à 5 ions lithium par unité TiINb:07.
Cela permet d'envisager une
très haute densité volumique d'énergie. La maille cristalline est monoclinique
avec un seul angle non
droit, a = B -- 90°, 7 = 120° et les paramètres de maille 4, b et c valent
respectivement 20,8; 12,2 et 3,8 À.
La maille conventionnelle est représentée figure 19.
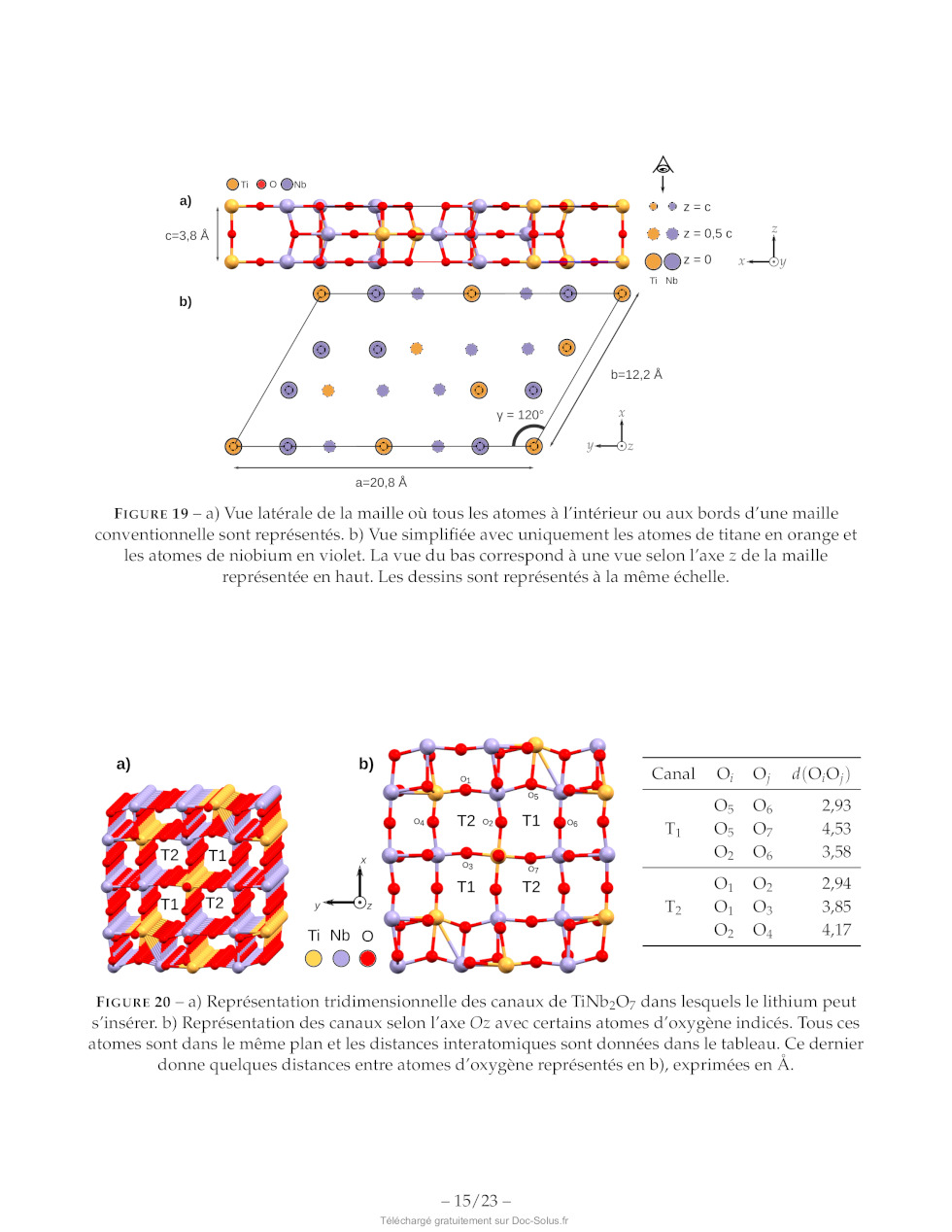
Dans cette structure, les polyèdres délimités par les atomes d'oxygène forment
deux types de canaux
selon l'axe Oz, appelés T1 et T2. Ils sont représentés sur la figure 20 et
quelques distances caractéristiques
sont également données. Ces canaux sont suffisamment grands pour que les ions
lithium puissent s'y
déplacer.
50. Donner la population (ou multiplicité) du motif TINb:07 pour la maille
représentée figure 19.
-- 14/23 -
OTi @ © @nb |
a=20,8 À
FIGURE 19 -- a) Vue latérale de la maille où tous les atomes à l'intérieur ou
aux bords d'une maille
conventionnelle sont représentés. b) Vue simplifiée avec uniquement les atomes
de titane en orange et
les atomes de niobium en violet. La vue du bas correspond à une vue selon l'axe
z de la maille
représentée en haut. Les dessins sont représentés à la même échelle.
Canal (@} O; d(O;O;)
O5 O6 2,93
T5 O5; O7 4,53
OO Ok 3,58
OL OO 2,94
D O1 O3 3,85
OO Ou 4,17
FIGURE 20 --- a) Représentation tridimensionnelle des canaux de TiNb:0; dans
lesquels le lithium peut
s'insérer. b) Représentation des canaux selon l'axe OZ avec certains atomes
d'oxygène indicés. Tous ces
atomes sont dans le même plan et les distances interatomiques sont données dans
le tableau. Ce dernier
donne quelques distances entre atomes d'oxygène représentés en b), exprimées en
À.
-- 15/23 -
51. Estimer la masse volumique de la maille représentée figure 19.
En 2019, une étude a identifié les sites d'insertion possibles pour le lithium
dans les différents canaux.
Trois sites d'insertion ont été caractérisés :
-- dans le canal T1 : un site d'insertion T11 où le lithium est au centre de la
cavité;
-- dans le canal T2 : deux sites d'insertion distincts appelés T28 et T2c.
Les chercheurs ont ensuite pu calculer l'énergie d'un ion lithium progressant
le long de chacun des
canaux selon l'axe OZ afin de les comparer (figure 21). L'extraction du lithium
étant difficile et son uti-
lisation dangereuse, ils ont également étudié d'autres cations pour faire des
batteries de nouvelle géné-
ration. Dans tous les cas, l'état de transition correspond au passage de
l'intérieur de la cavité aux plans
des atomes d'oxygène représentés figure 20 b).
a)
b)
LE (eV)
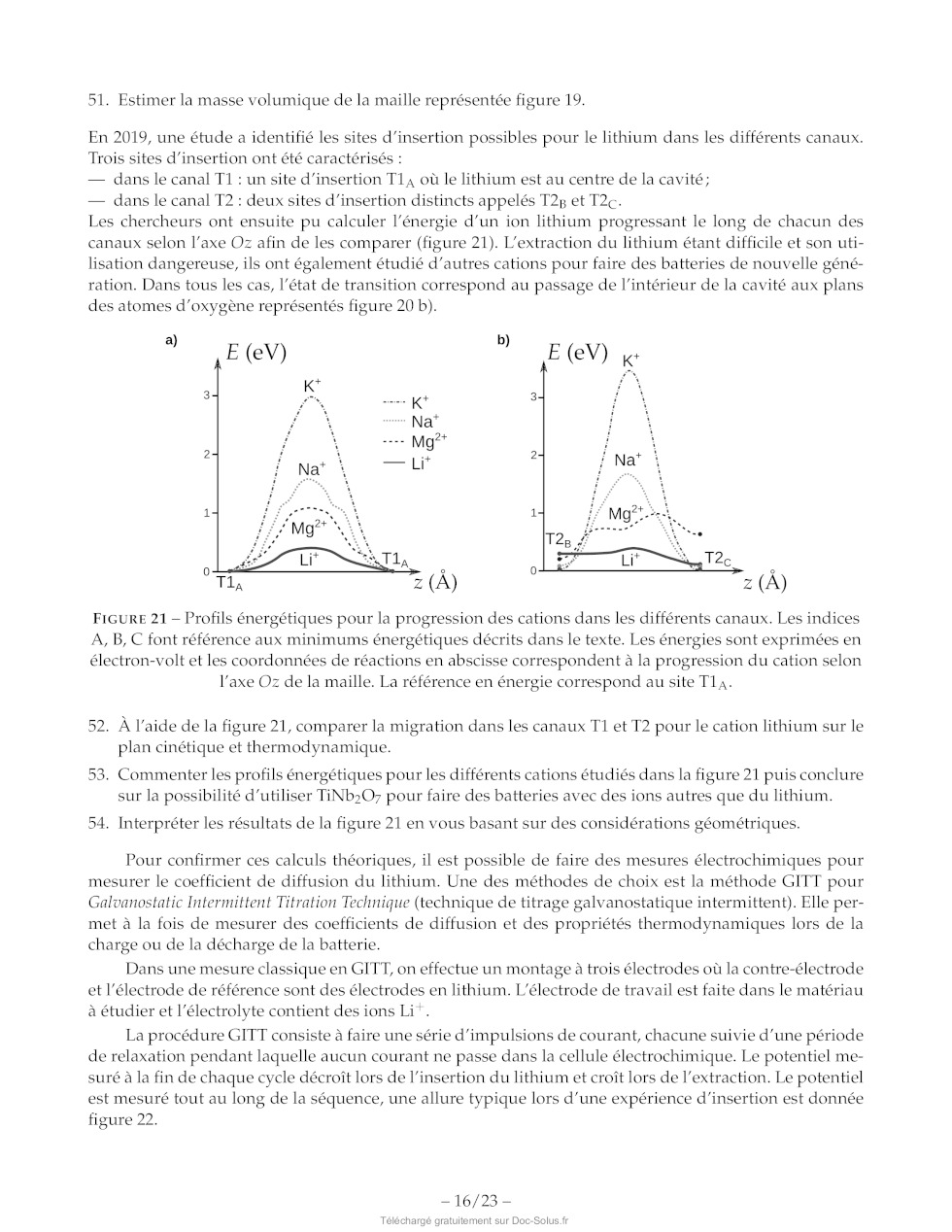
FIGURE 21 -- Profils énergétiques pour la progression des cations dans les
différents canaux. Les indices
À, B, C font référence aux minimums énergétiques décrits dans le texte. Les
énergies sont exprimées en
électron-volt et les coordonnées de réactions en abscisse correspondent à la
progression du cation selon
l'axe Oz de la maille. La référence en énergie correspond au site TA.
52. À l'aide de la figure 21, comparer la migration dans les canaux T1 et T2
pour le cation lithium sur le
plan cinétique et thermodynamique.
53. Commenter les profils énergétiques pour les différents cations étudiés dans
la figure 21 puis conclure
sur la possibilité d'utiliser TINb20; pour faire des batteries avec des ions
autres que du lithium.
54. Interpréter les résultats de la figure 21 en vous basant sur des
considérations géométriques.
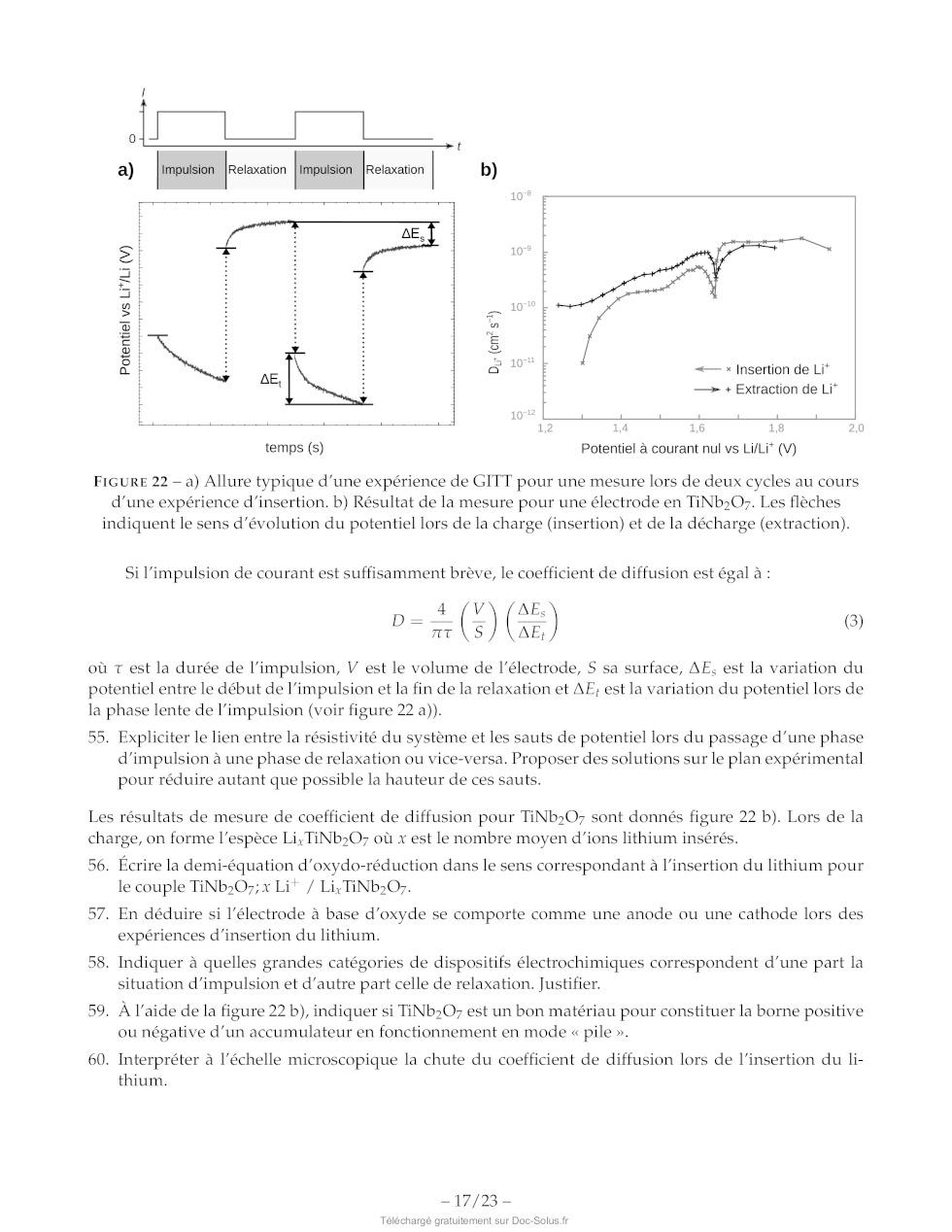
Pour confirmer ces calculs théoriques, il est possible de faire des mesures
électrochimiques pour
mesurer le coefficient de diffusion du lithium. Une des méthodes de choix est
la méthode GITT pour
Galvanostatic Intermittent Titration Technique (technique de titrage
galvanostatique intermittent). Elle per-
met à la fois de mesurer des coefficients de diffusion et des propriétés
thermodynamiques lors de la
charge ou de la décharge de la batterie.
Dans une mesure classique en GITT, on effectue un montage à trois électrodes où
la contre-électrode
et l'électrode de référence sont des électrodes en lithium. L'électrode de
travail est faite dans le matériau
à étudier et l'électrolyte contient des ions Li".
La procédure GITT consiste à faire une série d'impulsions de courant, chacune
suivie d'une période
de relaxation pendant laquelle aucun courant ne passe dans la cellule
électrochimique. Le potentiel me-
suré à la fin de chaque cycle décroît lors de l'insertion du lithium et croît
lors de l'extraction. Le potentiel
est mesuré tout au long de la séquence, une allure typique lors d'une
expérience d'insertion est donnée
figure 22.
-- 16/23 --
a) Impulsion [Relaxation |Impulsion [Relaxation b)
S Æ
5. :
+ F
1 |
u :
> 3
Z | a
Si 5
& | S <-- * Insertion de Li* e -- + Extraction de Li* temps (s) Potentiel à courant nul vs Li/Li* (V) FIGURE 22 --- à) Allure typique d'une expérience de GITT pour une mesure lors de deux cycles au cours d'une expérience d'insertion. b) Résultat de la mesure pour une électrode en TiNb:07. Les flèches indiquent le sens d'évolution du potentiel lors de la charge (insertion) et de la décharge (extraction). Si l'impulsion de courant est suffisamment brève, le coefficient de diffusion est égal à : 4 V AE. 7 (5) (Se:) 6) où t est la durée de l'impulsion, V est le volume de l'électrode, S sa surface, AE, est la variation du potentiel entre le début de l'impulsion et la fin de la relaxation et AE; est la variation du potentiel lors de la phase lente de l'impulsion (voir figure 22 à)). 95. Expliciter le lien entre la résistivité du système et les sauts de potentiel lors du passage d'une phase d'impulsion à une phase de relaxation ou vice-versa. Proposer des solutions sur le plan expérimental pour réduire autant que possible la hauteur de ces sauts. Les résultats de mesure de coefficient de diffusion pour TiNb:07 sont donnés figure 22 b). Lors de la charge, on forme l'espèce Li; TINb207 où x est le nombre moyen d'ions lithium insérés. 56. Écrire la demi-équation d'oxydo-réduction dans le sens correspondant à l'insertion du lithium pour le couple TiNb20O;; x Lit / Li, TINb:Oy. 57. En déduire si l'électrode à base d'oxyde se comporte comme une anode ou une cathode lors des expériences d'insertion du lithium. 58. Indiquer à quelles grandes catégories de dispositifs électrochimiques correspondent d'une part la situation d'impulsion et d'autre part celle de relaxation. Justifier. 59. À l'aide de la figure 22 b), indiquer si TINb2O> est un bon matériau pour
constituer la borne positive
ou négative d'un accumulateur en fonctionnement en mode « pile ».
60. Interpréter à l'échelle microscopique la chute du coefficient de diffusion
lors de l'insertion du li-
thium.
-- 17/23 --
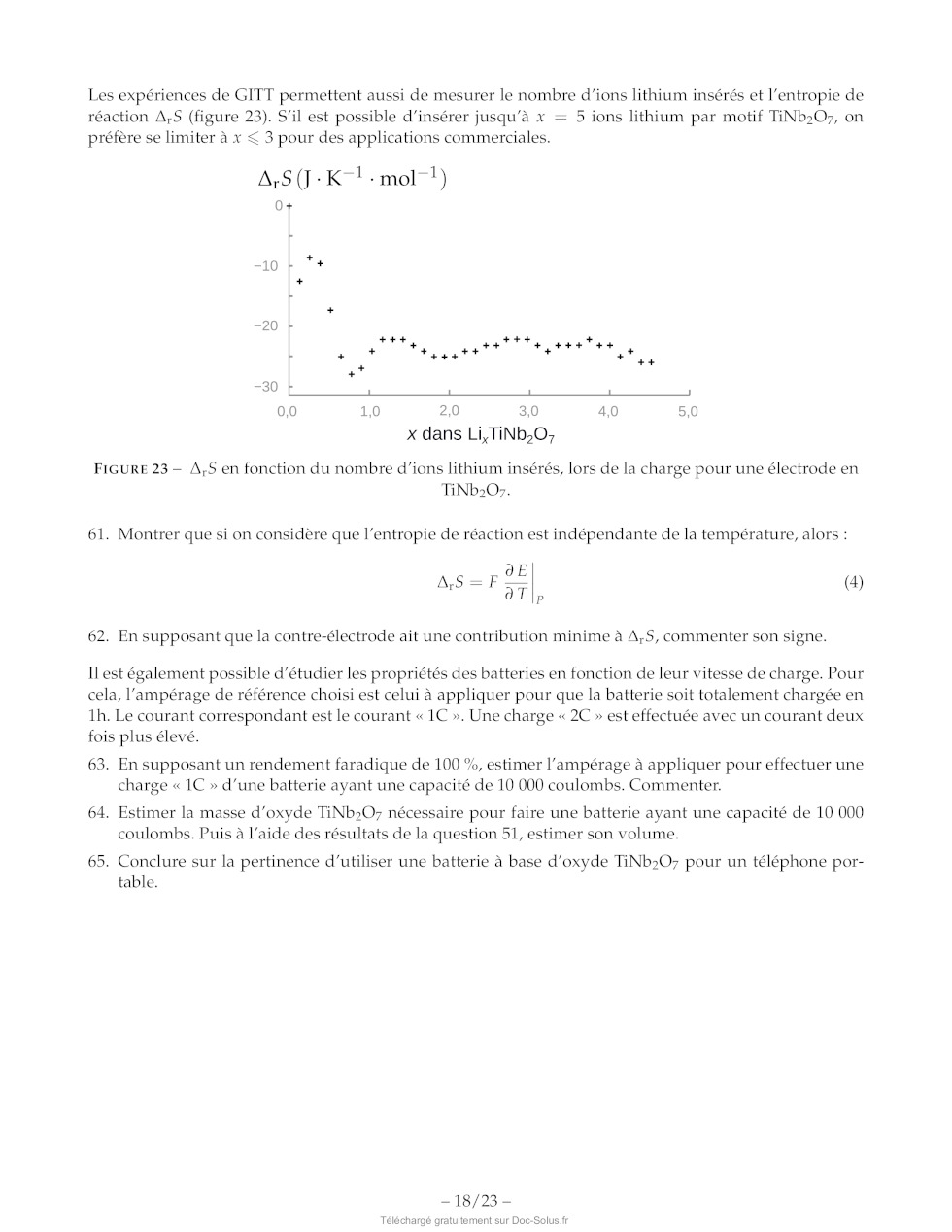
Les expériences de GITT permettent aussi de mesurer le nombre d'ions lithium
insérés et l'entropie de
réaction A;S (figure 23). S'il est possible d'insérer jusqu'à x = 5 ions
lithium par motif TiNb:07, on
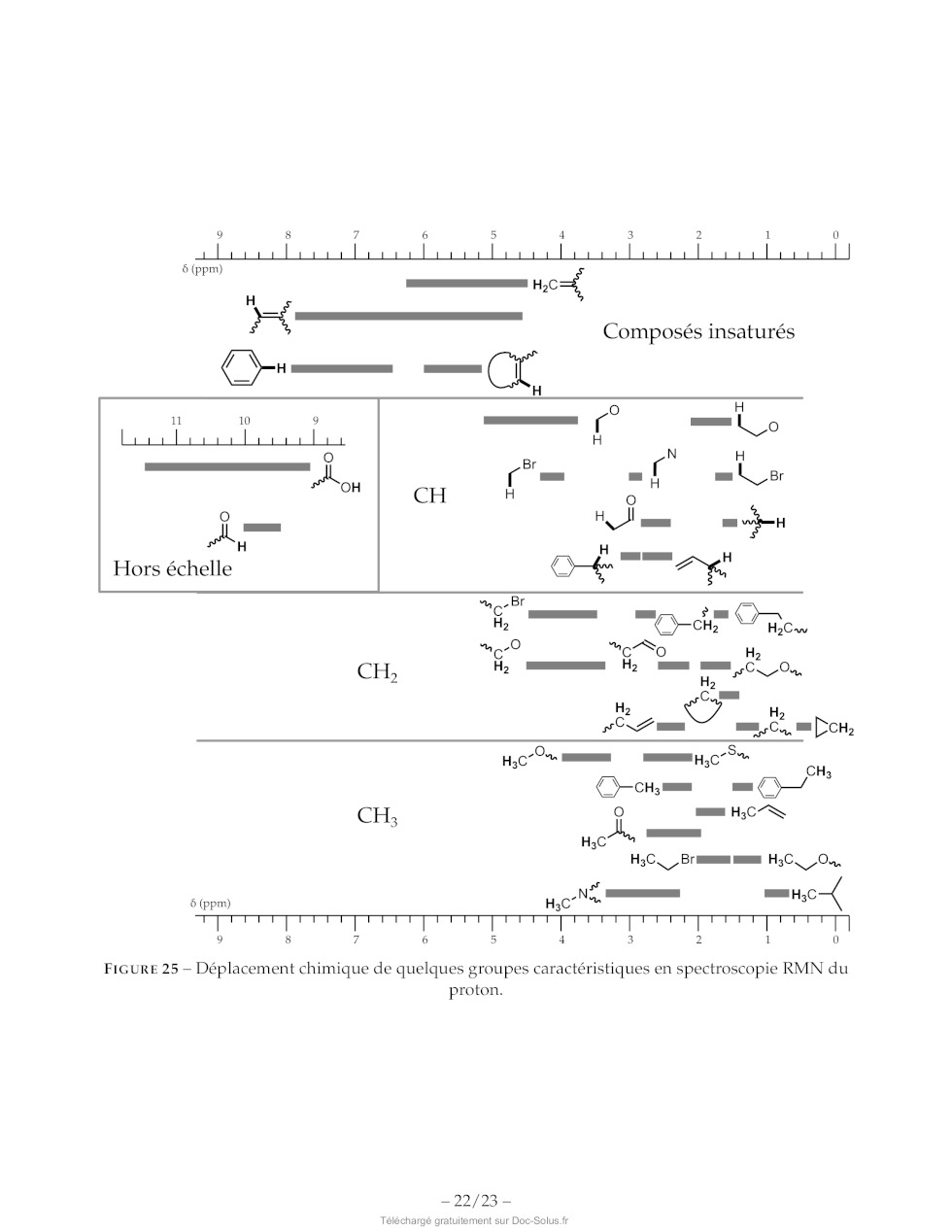
préfère se limiter à x < 3 pour des applications commerciales. A;S(T-K-l:mol 1) + x dans Li,TIND,0O; FIGURE 23 -- A,$ en fonction du nombre d'ions lithium insérés, lors de la charge pour une électrode en TiNb20O7. 61. Montrer que si on considère que l'entropie de réaction est indépendante de la température, alors : 0E AS =F-- 1 5T, (4) 62. En supposant que la contre-électrode ait une contribution minime à A,5, commenter son signe. Il est également possible d'étudier les propriétés des batteries en fonction de leur vitesse de charge. Pour cela, l'ampérage de référence choisi est celui à appliquer pour que la batterie soit totalement chargée en 1h. Le courant correspondant est le courant « 1C ». Une charge « 2C » est effectuée avec un courant deux fois plus élevé. 63. En supposant un rendement faradique de 100 %, estimer l'ampérage à appliquer pour effectuer une charge « 1C » d'une batterie ayant une capacité de 10 000 coulombs. Commenter. 64. Estimer la masse d'oxyde TiNb207 nécessaire pour faire une batterie ayant une capacité de 10 000 coulombs. Puis à l'aide des résultats de la question 51, estimer son volume. 65. Conclure sur la pertinence d'utiliser une batterie à base d'oxyde TINb207 pour un téléphone por- table. -- 18/23 - Abréviations DMF N,N-diméthylformamide A à reflux iPr groupement isopropyle Ms- mésyl ou méthylsulfonyl, CH3-50;- Ac- acétyle, CH3CO- anh. anhydre CI APTS acide paratoluènesulfonique o= No aq: aqueux NCS N-chlorosuccinimide tr Bn- benzyle, Ph-CH:- ' / Ph- Phényl, H5C4- Bz- benzoyle, Ph-CHO- ény?, HsCe Chbz- carboxybenzyl- substæch. substæoechiométrique DBU 1,8-diazabicyclo[5.4.0Jundéc-7-ène t.a. température ambiante DMAP 4-diméthylaminopyridine THE tétrahydrofurane Données -- Nombre d'Avogadro : N4 = 6,02 : 107 mol ! -- Constante de Faraday : F = 96500 C : mol"! --_ c0530°= sin60°= V? + 0,866 -- Z(Ni) = 28; Z(S) = 16; Z(B) =5 -- 1A--10 Um Élément O Ti Nb Masse molaire (g:mol !) 16,0 47,9 92,9 TABLEAU 6 -- Masse molaire de certains éléments. Ion O7 Lit Nat KY Mg AT Ga In Rayon (pm) 140 76 102 140 72 68 76 94 TABLEAU 7 -- Rayons ioniques en picomètres. -- 19/23 - Annexes Extrait de la documentation de la fonction numpy.log numpy.log(x) Logarithme naturel. Le logarithme naturel « log » est la fonction inverse de la fonction exponentielle, de manière à ce que log(exp(x)) = x. Le logarithme naturel est le logarithme de base « e ». Extrait de la documentation de la fonction numpy.polyfit numpy.polyfit(x, y, deg) Régression polynomiale par méthode des moindres carrés. Régression polynomiale avec un polynôme de la forme p(x) = plO] * x*xdeg + ... + pldeg] de degré « deg » par rapport aux points (x,y). Retourne les coefficients du polynôme p qui minimisent l'erreur quadratique. Paramètres : -- x: array_like, shape (M,) Abscisses des M points échantillonnés (x[i]l, y[il). -- y :array_like, shape (M,) or (M, K) Ordonnées des points échantillonnés. Il est possible de modéliser plusieurs jeux d'ordonnées partageant les mêmes abscisses en une seule fois en fournissant un tableau bidimensionnel qui contient un jeu de données par colonne. -- deg :int Degré du polynôme utilisé pour effectuer la régression. Retour : -- p:ñdarray, shape (deg + 1,) or (deg + 1, K) Coefficients du polynôme, avec le coefficient de plus haut degré en premier. Si y était bidimen- sionnel, les coefficients pour le K° jeu de données sont stockés dans p[L:,kl]. OH OH OH OH OH O O O O O NH NH NH: NH NH: -- OH SH N NH O O de OH histidine acide aspartique acide glutamique phénylalanine cystéine (1,70 : 6,04 :9,09) (1,95:3,71:9,66) (2,16:4,15 : 9,58) (2,18 ; 9,09) (1,91 : 8,14 : 10,28) OH OH OH OH OH O O O O O NH NH NH NH: NH: -- NH NE O O s, NH OH tryptophane asparagine glutamine tyrosine méthionine (2,38 ; 9,34) (2,16 ; 8,76) (2,18 ; 9,00) (2,24 ; 9,04 ; 10,10) (2,16 ; 9,08) FIGURE 24 -- Structure, nom et pK, successifs (entre parenthèses) de 10 acides aminés. -- 20/23 -- Liaison Groupe d'atomes Fonction Nombre d'onde Intensité caractéristique ou famille oc (cm |) O-H (libre) C-OH Alcool 3580-3670 Forte O-H C-OH Alcool 3200-3400 Forte (lié par liaison H) O-H COOH Acide carboxylique 3200-3400 Forte N-H C-NH- Amine, Amide 3100-3500 Moyenne C-N C=N Nitrile 2220-2260 Moyenne C-H Cycle benzénique Composés 3030-3080 Moyenne CH aromatiques Alcane 2810-3000 Forte Alcène 3000-3100 Moyenne C=O Carbonyle Aldéhyde, 1650-1730 Forte Cétone Carbonyle Acide 1680-1710 Forte CO-O-C Ester 1700-1740 Forte CO-N Amide 1650-1730 Forte C=C Alcène 1625-1680 Moyenne C-O Alcool, acide, 1050-1450 Forte ester C-C Alcane 1000-1250 Forte TABLEAU 8 -- Bandes caractéristiques en spectroscopie infrarouge. -- 21/23 - 9 8 7 6 5 4 3 2 1 0 iii tuuuutuurutuuuu trou tuuuutuurutruuutrurr lt] Ô (ppm) PR HO ' à Composés insaturés TA | RE CC H 11 10 9 Pa dé 0 uoiuluuuutuurulus H 0 r ou (= ={ = Los CH O # a L__| | Hors échelle Cr PS Br 5 REZ CZ H2 EUR Y-cH; HCav SI CH, H, mm H, mx un LC Ou, H C° sn & D + CS ==, C., = DcH; H cn ET sn 4," © cH 3 EUR Y--cH, mm mu / Y--/ CH; O mn HC H3C Br ER ER H3C. Ou, sf ( Ne, EE (ppm) H3C" H3C tt" 9 8 7 6 D 4 3 2 1 0 FIGURE 25 -- Déplacement chimique de quelques groupes caractéristiques en spectroscopie RMN du proton. -- 22/23 -- Fonction protégée Fonction alcool Groupement protecteur Conditions possibles pour la déprotection RLi RMgX KH LDA Acide Basique pH>12
CHO
X
X
X
61
X
Ac
BOM
X
R
R
X
R
61
66
X
Fonction
amine
TBS
Boc
Fmoc
Alloc
Ac
Bz
Bn
X
X
X
X
X
X
X
H2 , Pd/C
X
R
61
Bn
TMS
TBAF
Conditions usuelles de mis
du groupement protecteur
X
66
X
X
62
62
X
X
62
X
X
R
61
X
X
85% HCO2 H aq.,
60 °C, 1 h
Ac2 O, pyr, 20 °C, 12 h
BOMCl, (i-Pr)2 NEt,
20 °C, 12 h
BnBr, NaH, THF,
25 °C, 3 h
TMSCl, Et3 N, THF,
25 °C, 8 h
TBSCl, imidazole, DMF, 25 °C,
Boc2 O, THF, 40 °C, 24 h
FmocCl, NaHCO3 , dioxane aq.
AllocCl, pyr, 20 °C, 1 h
Ac2 O, pyr, 20 °C, 8 h
BzCl, pyr, 0 °C, 8 h
BnCl, K2 CO3 aq.,
100 °C, 30 min
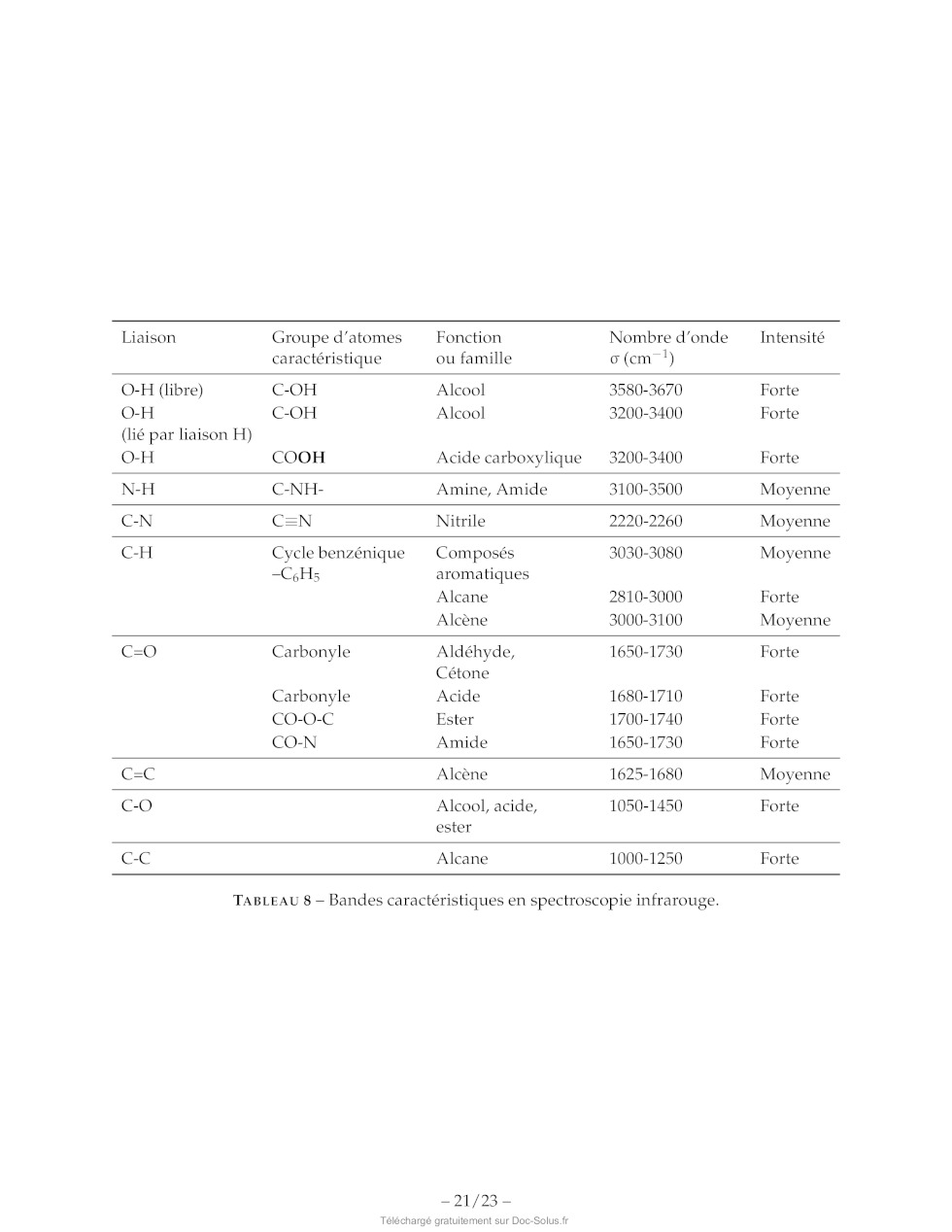
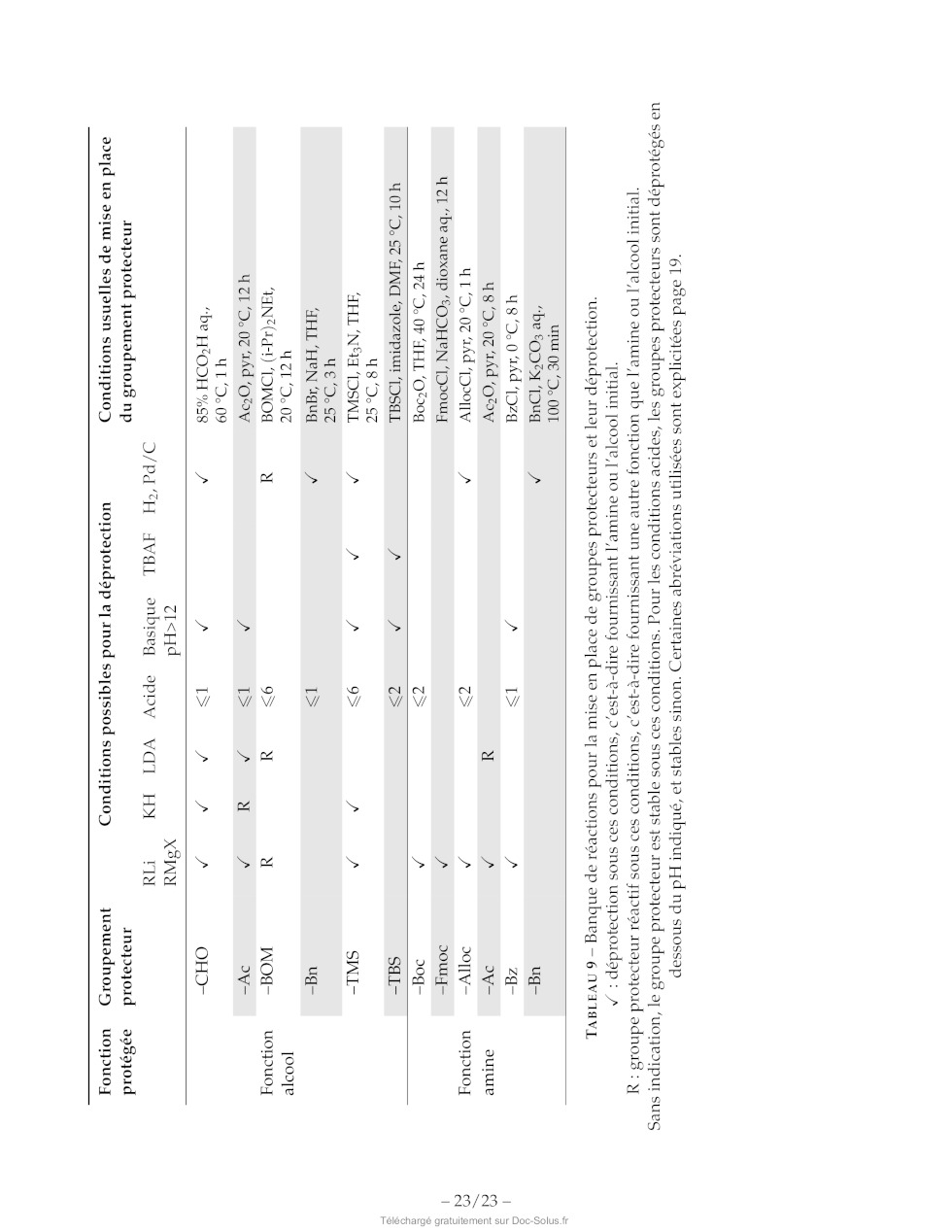
TABLEAU 9 Banque de réactions pour la mise en place de groupes protecteurs et
leur déprotection.
X : déprotection sous ces conditions, c'est-à-dire fournissant l'amine ou
l'alcool initial.
R : groupe protecteur réactif sous ces conditions, c'est-à-dire fournissant une
autre fonction que l'amine ou l'alcool init
Sans indication, le groupe protecteur est stable sous ces conditions. Pour les
conditions acides, les groupes protecteurs sont
dessous du pH indiqué, et stables sinon. Certaines abréviations utilisées sont
explicitées page 19.