
Mines Chimie PC 2025
| Thème de l'épreuve | Synthèse stéréosélective de la (+)-tubélactomicine. Application de la pervaporation à l'élimination d'eau. |
| Principaux outils utilisés | cristallographie, cinétique chimique, thermodynamique, mélanges binaires, chimie organique |
| Mots clefs | pervaporation, distillation |
Corrigé
:👈 gratuite pour tous les corrigés si tu crées un compte
👈 gratuite pour tous les corrigés si tu crées un compte
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
👈 gratuite pour ce corrigé si tu crées un compte
- - - - - - - - - - -
Énoncé complet
(télécharger le PDF)












Rapport du jury
(télécharger le PDF)





Énoncé obtenu par reconnaissance optique des caractères
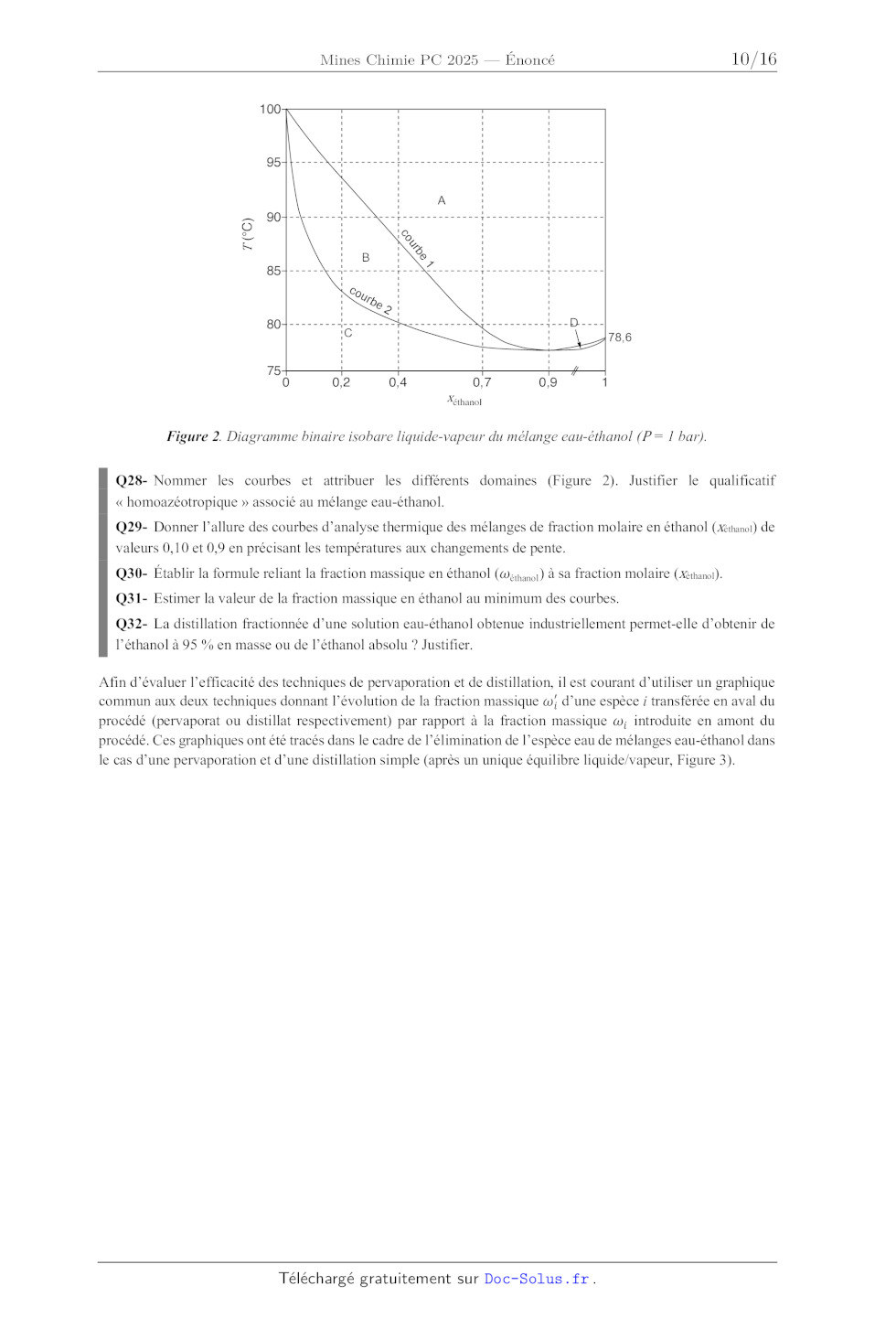
A2025 CHIMIE PC ÉCOLE NATIONALE DES PONTS et CHAUSSÉES, ISAE-SUPAERO, ENSTA PARIS, TÉLÉCOM PARIS, MINES PARIS, MINES SAINT-ÉTIENNE, MINES NANCY, IMT ATLANTIQUE, ENSAE PARIS, CHIMIE PARISTECH - PSL. Concours Mines-Télécom, Concours Centrale-Supélec (Cycle International). CONCOURS 2025 ÉPREUVE DE CHIMIE Durée de l'épreuve : 4 heures L'usage de la calculatrice ou de tout dispositif électronique est interdit. Les candidats sont priés de mentionner de façon apparente sur la première page de la copie : CHIMIE - PC L'énoncé de cette épreuve comporte 15 pages de texte. Si, au cours de l'épreuve, un candidat repère ce qui lui semble être une erreur d'énoncé, il le signale sur sa copie et poursuit sa composition en expliquant les raisons des initiatives qu'il est amené à prendre. . Les sujets sont la propriété du GIP CCMP. Ils sont publiés sous les termes de la licence Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de Modification 3.0 France. Tout autre usage est soumis à une autorisation préalable du Concours commun Mines Ponts. Ce problème comporte deux parties totalement indépendantes intitulées « Synthèse stéréosélective de la (+)-tubélactomicine A » et « Applications de la pervaporation à l'élimination d'eau ». Les données utiles à la résolution du problème sont fournies à la fin de l'énoncé (page 14). Pour l'écriture des mécanismes, le candidat pourra utiliser des notations simplifiées des molécules, lui permettant de se concentrer sur les groupes caractéristiques interagissant. Synthèse stéréosélective de la (+)-tubélactomicine A La (+)-tubélactomicine A (1) (Schéma 1) a été isolée de bouillons de culture d'une souche d'actinomycètes de Nocardia sp. MK703-102F1 et a montré une activité antimicrobienne forte et spécifique contre Mycobacterium sp.1 Sa structure, qui a été déterminée par cristallographie de diffraction aux rayons X, est constituée d'une macrolactone (ester cyclique) à 16 chaînons fusionnée avec un squelette trans-décaline.2 CO2H HO CO2H 18 16 HO 23 O 14 12 11 1 2 H + 16 O H 3 8 I 23 14 4 O 1 18 6 SnBu3 2 12 11 2 OH OSEM H 3 8 H 3 4 6 OH OH SEM = (CH3)3SiCH2CH2OCH2 (+)-Tubélactomicine A (1) Schéma 1. Approche rétrosynthétique de la (+)-tubélactomicine A. Une synthèse totale de la (+)-tubélactomicine A (1) a été décrite par l'équipe de Tadano et repose sur l'assemblage du précurseur 2 de la partie macrolactone avec l'iodure vinylique 3, possédant un motif trans-décaline (Schéma 1).3 Dans la suite, seule la synthèse du précurseur 3 sera étudiée. Q1- Donner la configuration absolue des centres stéréogènes 2 et 3 de la (+)-tubélactomicine A (1) en justifiant votre réponse. L'iodure vinylique 3 a été préparé à partir de l'aldéhyde 4 et du phosphonate 5 (Schéma 2), dont nous allons détailler les synthèses. 1 M. Igarashi, C. Hayashi, Y. Homma, S. Hattori, N. Kinoshita, M. Hamada, T. Takeuchi J Antibiot. 2000, 53, 10961101. M. Igarashi, H. Nakamura, H. Naganawa, T. Takeuchi J Antibiot. 2000, 53, 11021107. 3 (a) T. Motozaki, K. Sawamura, A. Suzuki, K. Yoshida, T. Ueki, A. Ohara, R. Munakata, K.-I. Takao, K.-I. Tadano Org. Lett. 2005, 7, 22612264. (b) T. Motozaki, K. Sawamura, A. Suzuki, K. Yoshida, T. Ueki, A. Ohara, R. Munakata, K.-I. Takao, K.I. Tadano Org. Lett. 2005, 7, 22652267. 2 Page 1/15 I O OTBS OSEM H O O 3 H 4 OH TMS O P(OEt)2 + O TMS = (CH3)3Si TBS = tBu(CH3)2Si 5 Ph Schéma 2. Préparation de l'intermédiaire 3. Synthèse de l'aldéhyde 4 La synthèse stéréosélective de l'aldéhyde 4 a été réalisée, en plusieurs étapes, à partir d'une source de chiralité commerciale, l'acide (R)-malique (6) (Schéma 3). HO2C OH EtOH CO2H H SO 2 4 reflux 1) LDA (2 équiv), THF EtO2C CO2Et OH acide (R)-malique (6) 8 réactif 9 HO OH 10 2) EtO2C Br 3) hydrolyse 7 OH PhCH(OMe)2 Amberlyst 15 CH2Cl2 réactif 12 HO O 11 CO2Et OH 8 BnO O O O 13 Ph Ph OTBS 13 1) BH3·Me2S 2) H2O2, NaOH aq. réactif 15 14 OTBS réactif 17 BnO O 16 O HO O 18 Ph O Ph OTBS 18 réaction O O 4 LDA = O N Li Ph Schéma 3. Synthèse de l'aldéhyde 4 à partir de l'acide (R)-malique (6). L'acide (R)-malique (6) est dans un premier temps estérifié à l'aide d'éthanol en excès en utilisant le protocole suivant : à une solution d'acide (R)-malique (6) (6,7 g) dans l'éthanol (300 mL) sont ajoutés1,4 mL d'acide sulfurique à 99,8 %. Le mélange est chauffé au reflux pendant 2 heures, concentré sous vide puis purifié par chromatographie sur gel de silice pour donner 8,6 g du diester 7 sous forme d'une huile incolore. Q2- Proposer un mécanisme pour la réaction d'estérification d'un acide carboxylique (noté RCO2H) par de l'éthanol en milieu acide. Q3- Calculer le rendement obtenu dans cette première étape. Quelles sont les conditions utilisées qui permettent d'optimiser le rendement de cette réaction ? Q4- Proposer une méthode pour préparer le diisopropylamidure de lithium (LDA) à partir du n-butyllithium (nBuLi). Pourquoi n'a-t-on pas fait réagir directement du n-butyllithium sur le composé 7 ? Q5- Donner la structure de l'intermédiaire formé par action du diisopropylamidure de lithium (LDA) sur le substrat 7 et justifier la quantité de LDA utilisée. Q6- Proposer un mécanisme pour la formation de 8 à partir de 7. Page 2/15 Q7- Sachant que les cations lithium peuvent chélater deux oxygènes différents d'une même molécule afin de former un cycle rigide (ici à 6 atomes) comme indiqué sur la figure ci-contre, justifier la diastéréosélectivité observée lors de la formation du composé 8 et donner la structure du stéréoisomère minoritairement formé. Li Li O O Q8- Proposer des conditions réactionnelles (réactif 9) pour effectuer la transformation du diester 8 en triol 10. L'acétalisation du triol 10 en hydroxyacétal 11 est effectuée en utilisant du (diméthoxyméthyl)benzène et de l'Amberlyst 15, une résine échangeuse d'ions capable de fournir des protons. Le spectre RMN du proton du produit 11 (300 MHz, CDCl3) présente les signaux suivants (d en ppm)4 : 1,602,42 (3H, m), 3,704,00 (5H, m), 4,20 (1H, s large), 5,05 (1H, d, J = l0,2 Hz), 5,06 (1H, d, J = 17,1 Hz), 5,48 (1H, s), 5,79 (1H, ddt, J = 17,1 ; 10,2 et 7,0 Hz), 7,15-7,55 (5H, m). g (s = singulet ; d = doublet ; t = triplet ; m = multiplet) H h' f Q9- A l'aide des tables (Annexe 5 page 15), attribuer le plus précisément possible l'ensemble des signaux du spectre RMN 1H du composé 11 en utilisant la numérotation proposée. Justifier soigneusement les attributions. a b H iO c d Hh O e O 11 Ph Q10- Proposer des réactifs (réactif 12) permettant la transformation de 11 en 13 (voir Document 1). Document 1. Protection de la fonction alcool via la formation d'éthers. Dans une synthèse, les groupements hydroxy sont, la plupart du temps, protégés. Parmi les groupements protecteurs couramment utilisés, on rencontre les éthers de benzyle (ROBn) ou les éthers silylés (ROSiR1R2R3) qui sont formés via des réactions de substitution. Br R OH NaH R1 R2 Si Cl R3 R OH Et3N Pd/C cat. R O = ROBn éther de benzyle R1 R O Si R2 R3 F H2 R OH R OH éther silylé Les éthers de benzyle sont facilement déprotégés par hydrogénolyse (H2, Pd/C cat.), alors que les éthers silylés sont retirés en présence d'ions fluorures (Bu4NF), permettant de régénérer la fonction alcool. Parmi les éthers silylés courants on rencontre le triméthylsilyle (R1 = R2 = R3 = Me), le tert-butyldiméthylsilyle (R1 = tBu, R2 = R3 = Me) abrégé TBS ou le tert-butyldiphénylsilyle (R1 = tBu, R2 = R3 = Ph) abrégé TBDPS. Le composé 13 est ensuite transformé en un intermédiaire 14 selon le mode opératoire suivant : sous atmosphère d'argon, le composé 13, en solution dans le THF anhydre, est traité par BH3·Me2S et agité pendant 2,5 heures à température ambiante avant d'ajouter un mélange de soude et de peroxyde d'hydrogène. Après une heure de réaction, une solution à 10 % de thiosulfate de sodium est ajoutée à 0 °C avant traitement et purification pour donner le composé 14 avec un rendement de 86 %. Q11- Donner la structure du composé 14 formé et justifier la régiosélectivité de la réaction. Quel est l'intérêt d'ajouter une solution de thiosulfate de sodium à la fin de la réaction ? Q12- Proposer les réactifs (réactif 15 et réactif 17) permettant la transformation de 14 en 18. Q13- Quel type de réaction permet la transformation de l'alcool 18 en aldéhyde 4 ? 4 Y. Morimoto, A. Mikami, S.-I. Kuwabe, H. Shirahama Tetrahedron: Asymmetry 1996, 7, 33713390. Page 3/15 Synthèse du phosphonate 5 Le composé 4 ainsi formé a été mis en réaction avec le phosphonate 5, dont la synthèse a été réalisée à partir du malonate 19 (Schéma 4). La synthèse du phosphonate 5 débute par l'action de l'hydrure de sodium sur le malonate 19 en présence de triiodométhane dans l'éther diéthylique au reflux. Un composé 20, de formule brute C9H14I2O4, est obtenu avec un rendement de 65 %. La réaction de ce composé 20 avec de l'hydroxyde de potassium dans un mélange hydroalcoolique au reflux permet la formation d'un intermédiaire ionique instable [21] qui se décompose rapidement avec dégagement gazeux. L'iodure vinylique 22 est ensuite obtenu après acidification du milieu réactionnel. CO2Et CO2Et 19 Et2O, reflux 20 (C9H14I2O4) TMS 23 I I CHI3, NaH [21] 23 TMS OH réactif 25 24 OH CO2H 22 TMS Pd(PPh3)4 cat. CuI cat. TMS KOH EtOH, H2O reflux LiAlH4 Br 26 O P(OEt)2 P(OEt)3 reflux 5 NH Schéma 4. Synthèse du phosphonate 5. Q14- Donner la structure du composé 20 formé lors de la première étape et indiquer le type de réaction qui a permis son obtention. Q15- Donner la structure de l'intermédiaire ionique instable [21] obtenu à partir du composé 20. Proposer un mécanisme permettant d'expliquer sa formation. Q16- Proposer un mécanisme réactionnel pour l'obtention du composé 22 à partir de l'intermédiaire ionique [21]. Dans la suite de la synthèse, l'alcool allylique 23, obtenu par réduction de 22, est mis en réaction avec le triméthylsilylacétylène via une réaction de Sonogashira, catalysée par le palladium, pour conduire au composé 24 (Schéma 4). Le mécanisme de cette réaction de Sonogashira peut être décrit via un cycle catalytique simplifié faisant intervenir quatre étapes (Schéma 5). Page 4/15 PdL4 HO 2L I PdL2 OH 24 TMS D A L HO 23 OH I L = PPh3 Pd Pd L L L TMS B CuI cat. NH C I I L, OH H NH TMS H L Pd L TMS Schéma 5. Cycle catalytique simplifié de la réaction de Sonogashira. Q17- Donner les noms des étapes A, B et D du cycle catalytique. Indiquer la variation du nombre d'oxydation formel du centre métallique pour chacune de ces étapes. L'alcool 24 obtenu est alors transformé en bromure allylique 26, qui permet de conduire au phosphonate 4 par réaction au reflux du phosphite de triéthyle (P(OEt)3) (Schéma 4). Q18- Proposer deux méthodes différentes (réactif 25) pour transformer l'alcool allylique 24 en bromure allylique 26. Quelles seraient les conditions les plus adaptées dans le cas de cette synthèse ? Synthèse de l'iodure vinylique 3 L'aldéhyde 27 a été obtenu à partir de l'aldéhyde 4 et du phosphonate 5 par une suite de réactions qui ne sont pas détaillées ici (Schéma 6). Cet aldéhyde 27 a ensuite été transformé en iodure vinylique 3 en quelques étapes que nous allons étudier. Il subit dans un premier temps une réaction d'oxydation, en utilisant du chlorite de sodium NaClO2, pour conduire à l'acide carboxylique 28 qui est ensuite transformé en diol 29 en trois étapes, l'une de ces trois étapes étant une hydrolyse acide de l'acétal présent dans le composé 28 par l'acide éthanoïque aqueux. Page 5/15 OTBS O O 4 O P(OEt)2 + O CHO H TMS TMS H 5 Ph O 27 O Ph CO2H H TMS 27 CO2SEM H NaClO2, NaH2PO4 2-méthylbut-2-ène tBuOH, H2O H 28 O H O 29 OH OH Ph 29 TsCl (1 éq) Et3N, DMAP CH2Cl2 30 I CO2SEM H NaBH4 O DMSO, 100 °C H 31 OH 3 SEM = (CH3)3SiCH2CH2OCH2 TsCl = OSEM H O S Cl O DMAP = Me2N N DMSO = H OH O S Schéma 6. Fin de la synthèse de l'iodure vinylique 3. Q19- Proposer une structure de Lewis de l'ion chlorite ClO2 . Q20- Expliquer pourquoi un mélange des deux solvants tert-butanol/eau est employé lors de la réaction de formation de 28. Q21- Proposer un mécanisme pour l'hydrolyse en milieu acide de la fonction acétal du composé 28 (on utilisera une notation simplifiée pour 28). L'alcool primaire du diol 29 est ensuite réduit en alcane en deux étapes pour conduire au composé 31 qui est finalement transformé en iodure vinylique 3 (Schéma 6). Q22- Donner la structure du composé 30. Expliquer la régiosélectivité de cette réaction. Q23- Quel type de réaction permet d'obtenir le composé 31 à partir du composé 30 ? Aurait-on pu utiliser de l'hydrure de sodium à la place du borohydrure de sodium ? Justifier. Finalement, la (+)-tubélactomicine A (1) a été synthétisée, en quelques étapes, qui ne seront pas étudiées ici, par couplage, catalysé par le palladium, des composés 2 et 3 (Schéma 1, page 1) suivi de réactions de fonctionnalisation. Page 6/15 Applications de la pervaporation à l'élimination d'eau La pervaporation est une alternative complémentaire à la distillation, qui est notamment bien adaptée à l'extraction d'un composé minoritaire. Elle permet alors une économie d'énergie importante par rapport à la distillation. Nous allons nous intéresser à l'application de la pervaporation dans deux procédés visant l'élimination d'eau dans un mélange : l'obtention d'éthanol absolu à partir d'un mélange eau-éthanol et le déplacement d'un équilibre d'estérification. Développée depuis les années 1960 à la fois aux USA et en Europe, la pervaporation est historiquement un procédé de séparation de mélanges liquides ; elle met à profit le transfert sélectif de matière à travers une membrane polymère organique ou inorganique de type zéolithe.5 Au cours de cette opération, le flux de matière qui traverse la membrane est vaporisé puis récupéré en aval, en général par condensation sur une paroi froide : le liquide obtenu est appelé splitéat ou pervaporat. Le liquide resté en amont est enrichi en composé ayant le moins diffusé au travers de la membrane : il est appelé rétentat. Alimentation Compartiment sous vide Paroi froide (condensation) Membrane Rétentat (riche en ) Espèce la moins splitéable Espèce la plus splitéable Perméat ou pervaporat (riche en ) Figure 1. Schéma de principe de la pervaporation. Par rapport à la distillation, la pervaporation est particulièrement intéressante pour la séparation de composés ayant des volatilités voisines. En effet, alors que la distillation demande dans ce cas un nombre important de plateaux et un taux de reflux élevé entraînant une forte consommation énergétique liée aux vaporisations successives, la pervaporation permet, en un seul étage, de séparer majoritairement l'un des composés par vaporisation à travers la membrane, de préférence le composé minoritaire car la densité de flux est faible comparativement aux autres procédés membranaires. Membrane de type zéolithe pour la pervaporation Les zéolithes sont des aluminosilicates solides poreux appelés également tamis moléculaires : les molécules passant au travers des pores de la zéolithe peuvent être séparées selon leur taille, les plus petites progressant plus rapidement dans la membrane. Le flux de matière dépend du volume poreux, qui correspond en moyenne à 45% du volume de la zéolithe. Nous nous intéressons plus particulièrement à une zéolithe de formule Si96O192 par maille, dont les paramètres de maille ont été déterminés (Tableau 1). 5 C. Castel, E. Favre, S. Rode, D. Roizard, E. Carretier, C. Arnal-Hérault, R. Clément, A. Jonquières Techniques de l'Ingénieur 2020, J2820 v2, 127. Page 7/15 Tableau 1. Paramètres de maille de la zéolithe de formule Si96O192. Côtés (pm) Angles (°) a b c a b g 2000 2000 1300 90 90 90 Q24- Calculer la masse volumique de cette zéolithe (en g·cm3). Q25- Calculer le volume poreux Vp, exprimé usuellement en cm3·g1 de la zéolithe. À partir du volume poreux et d'analyses cristallographiques, il a été possible de déterminer le diamètre des pores les plus grands de différentes zéolithes (Tableau 2). Tableau 2. Valeurs du diamètre des pores les plus grandes d'une série de zéolithes. Zéolithe Diamètre des pores (pm) ZMS-5 560 Chabazite 380 Zéolithe Y 740 Zéolithe BETA 770 Q26- À partir des données en annexe, estimer la taille approximative d'une molécule d'eau (pour simplifier les calculs, on considèrera l'eau comme une molécule linéaire). Q27- Quelle(s) zéolithe(s) serai(en)t efficace(s) pour séparer un mélange eau-éthanol par pervaporation sur membrane zéolithe, sachant que la taille d'une molécule d'éthanol est de 0,47 nm ? Application de la pervaporation à l'obtention d'éthanol absolu5 La synthèse industrielle de l'éthanol conduit à l'obtention d'un mélange eau-éthanol de pourcentage massique allant de 10 à 25 % en éthanol suivant le procédé de synthèse. Pour obtenir de l'éthanol commercialisable, il est nécessaire de déshydrater la solution jusqu'à obtenir deux conditionnements usuels : l'éthanol à 95 % (en masse) et l'éthanol absolu (99,7 % en masse d'éthanol minimum). Dans un premier temps nous allons nous intéresser à la déshydratation de l'éthanol par distillation (Figure 2). Page 8/15 100 95 co ur B be T (°C) A 90 1 85 co 80 75 urb e2 D C 0 0,2 78,6 0,4 0,7 xéthanol 0,9 1 Figure 2. Diagramme binaire isobare liquide-vapeur du mélange eau-éthanol (P = 1 bar). Q28- Nommer les courbes et attribuer les différents domaines (Figure 2). Justifier le qualificatif « homoazéotropique » associé au mélange eau-éthanol. Q29- Donner l'allure des courbes d'analyse thermique des mélanges de fraction molaire en éthanol (xéthanol) de valeurs 0,10 et 0,9 en précisant les températures aux changements de pente. Q30- Établir la formule reliant la fraction massique en éthanol ($éthanol ) à sa fraction molaire (xéthanol). Q31- Estimer la valeur de la fraction massique en éthanol au minimum des courbes. Q32- La distillation fractionnée d'une solution eau-éthanol obtenue industriellement permet-elle d'obtenir de l'éthanol à 95 % en masse ou de l'éthanol absolu ? Justifier. Afin d'évaluer l'efficacité des techniques de pervaporation et de distillation, il est courant d'utiliser un graphique commun aux deux techniques donnant l'évolution de la fraction massique $!" d'une espèce i transférée en aval du procédé (pervaporat ou distillat respectivement) par rapport à la fraction massique $! introduite en amont du procédé. Ces graphiques ont été tracés dans le cadre de l'élimination de l'espèce eau de mélanges eau-éthanol dans le cas d'une pervaporation et d'une distillation simple (après un unique équilibre liquide/vapeur, Figure 3). Page 9/15 1,0 pervaporatio n 0,8 ` eau 0,6 0,4 ELV 0,2 0 0 0,2 0,4 eau 0,6 0,8 1,0 Figure 3. Fraction massique en eau %"#$% en sortie du dispositif dans la 1ère vapeur extraite pour la distillation (courbe du bas) et dans le pervaporat pour la pervaporation (courbe du haut) en fonction de la fraction massique en eau %#$% dans la solution eau-éthanol initiale. Q33- Par les informations qu'elle apporte, à quelle courbe du diagramme binaire eau-éthanol s'apparente la courbe ELV (Équilibre Liquide-Vapeur, Figure 3) ? " Q34- Déterminer la fraction massique en éthanol $é'()*+,(.) de la première vapeur issue de la distillation d'une solution initiale à 25 % en éthanol. À partir de la courbe ELV (Figure 3), il est aussi possible de suivre la composition de la vapeur lors d'équilibres liquide/vapeur successifs dans une colonne à distiller. " Q35- En considérant que la vapeur de composition $é'()*+,(.) précédente se recondense et qu'un nouvel " équilibre liquide/vapeur s'établit dans la colonne, déterminer la fraction massique $é'()*+,(0) en éthanol de cette nouvelle vapeur. Q36- Une opération similaire peut-être réalisée en plusieurs fois jusqu'à ce que le système atteigne l'homoazéotrope. En faisant appel aux caractéristiques d'un homoazéotrope, indiquer comment retrouver sa composition massique sur le graphique (Figure 3). Pour une même solution eau-éthanol initiale à 25 % en éthanol, la pervaporation donne un résultat différent de la distillation. Q37- À partir du graphique (Figure 3), montrer qualitativement que la pervaporation permet d'éliminer plus efficacement l'eau du mélange considéré que la distillation. Q38- À partir de la Figure 4, déterminer le débit en entrée &1,! et le débit du pervaporat &1,345 qui ont été nécessaires pour obtenir les fractions massiques affichées. Page 10/15 Mélange initial eau,i = 75 % Dm,i = ? Membrane Rétentat eau,rét = 0,3 % Dm,rét = 2,5 kg·h1 Pervaporat eau,per = 98 % Dm,per = ? Figure 4. Caractéristiques en débit massique (Dm) et fraction massique de la pervaporation d'un mélange eau-éthanol. Q39- Conclure quant à la composition de l'éthanol commercialisable par pervaporation de la solution initiale : éthanol à 95% et/ou absolu ? Application de la pervaporation à un déplacement d'équilibre6 La pervaporation est une alternative intéressante à la distillation historiquement utilisée pour déplacer les équilibres de distillation (comme le Dean-Stark), tant en termes de nombre d'étapes de traitement qu'en coût énergétique. Cette technique a été mise en oeuvre pour le déplacement de l'équilibre d'estérification de l'acide propanoïque par le propan-1-ol. Q40- Écrire l'équation de la réaction d'estérification entre l'acide propanoïque et le propan-1-ol en formule topologique. Q41- Déterminer la valeur de la constante d'équilibre à 298 K de cette réaction à partir des données thermodynamiques fournies en annexe (p 14). Q42- Montrer que l'élimination d'eau est une stratégie intéressante pour favoriser la formation d'ester. La réponse sera argumentée en faisant appel à la notion de quotient réactionnel. Dans le cadre d'une étude cinétique,6 l'estérification est modélisée par deux actes élémentaires de sens opposés, de constantes de vitesse (. dans le sens direct et (6. dans le sens opposé. Les ordres partiels sont égaux à 1 pour l'alcool, l'acide carboxylique, l'eau et l'ester. La réaction se fait en présence d'un catalyseur (noté Cat), dont l'ordre partiel, noté ,, caractérise la cinétique autant de l'étape directe que de l'étape indirecte. Les réactifs alcool et acide carboxylique sont introduits dans les proportions stoechiométriques. Q43- Exprimer la vitesse volumique - de formation de l'ester en fonction de la concentration en acide carboxylique, en alcool, en ester, en eau, en catalyseur et des constantes de vitesse (. et (6. . Afin de déterminer l'ordre partiel par rapport au catalyseur, différentes expériences ont été menées en faisant varier uniquement la concentration en catalyseur (Figure 5). 6 M-O. David, Déplacement d'une réaction chimique par pervaporation, Thèse de l'INPL, octobre 1991. Page 11/15 vitesse (mol·L1·h1) 0,4 x x 0,3 0,2 x x x x 0,1 x 0,0 0,0 0,1 0,2 0,3 [Cat] (mol·L1) Figure 5. Variation de la vitesse de formation de l'ester avec la concentration en catalyseur. Q44- Estimer l'ordre partiel par rapport au catalyseur. La température impacte la cinétique de la réaction d'estérification. Dans le cadre de la réaction d'estérification étudiée, l'évolution du logarithme de la constante de vitesse (. en fonction de la température peut être modélisée par l'expression : 5700 + 10 5 avec T la température en Kelvin. ln (. = - Q45- Déterminer l'énergie d'activation de la réaction d'estérification. Q46- En déduire que la loi cinétique globale de l'estérification répond à l'équation différentielle (1) : 9: ,7 ,7 = (. (1 - :)0 [@A;] - (6. : 0 [@A;] 9; < < (D) où X est le taux de conversion de l'acide carboxylique, ,7 la quantité initiale d'acide carboxylique et V le volume du milieu réactionnel (considéré comme constant). Lorsque l'estérification est menée en amont d'une membrane de pervaporation, qui permet d'extraire l'eau du milieu réactionnel, ce dernier peut être considéré comme étant le rétentat. Au bilan, sur un intervalle de temps dt, l'eau formée lors de l'estérification 9,4)8 :+51é4 se répartit de la façon suivante : une partie 9,4)8,5é'4*')' s'accumule dans le rétentat et l'autre partie traverse la membrane avec un flux G dont l'étude montre qu'il est proportionnel à la quantité d'eau dans le rétentat : G = A. ,4)8,5é'4*')' avec G en mol·s1. Q47- Montrer que la vitesse d'évolution globale de la quantité d'eau dans le rétentat au cours de l'estérification suit la loi cinétique (2) : 9I 9: = -AI 9; 9; (J) où X est le taux de conversion de l'acide carboxylique et I le rapport de la quantité d'eau dans le rétentat sur la quantité initiale d'acide carboxylique (I = ,4)8,5é'4*')' /,+ ). La résolution numérique du système formé des équations (1) et (2) permet d'obtenir l'évolution au cours du temps du taux de conversion en ester et du rapport de la quantité en eau dans le rétentat sur la quantité initiale d'acide (Figure 6). Page 12/15 1,0 Évolution de X et Y x Ester (X) 0,8 xx x x x x x+ 0,4 x++ + + + + x+ 0,6 0,2 0,0 Eau (Y) x 0 + 10 20 30 Temps (h) Conditions expérimentales : mélange stoechiométrique des réactifs, APTS à 1 % en masse dans le mélange, T = 50 °C. Figure 6. Comparaison entre les points expérimentaux et les courbes théoriques donnant l'évolution du taux de conversion en ester (X) et du rapport de la quantité en eau dans le rétentat sur la quantité initiale d'acide (Y) en fonction du temps. Q48- Quel processus, de l'estérification ou de la pervaporation, impacte les premiers instants de l'évolution de la quantité d'eau ainsi que la fin de la courbe ? Justifier. Q49- Estimer le rendement final en ester. Conclure sur l'efficacité de la pervaporation dans l'optimisation du rendement d'une estérification. Fin de l'épreuve Page 13/15 Annexes Annexe 1. Constantes usuelles et approximation de calculs. Constante d'Avogadro : L; 6.1023 mol1. Constante des gaz parfaits M 8 J·K1·mol1. Il sera considéré que N.7/= 4. Annexe 2. Données thermodynamiques (considérées indépendantes de la température). Espèce Propan-1-ol() Acide propanoïque() Propanoate de propyle() H2 O() : U7 (kJ·mol1) 490 280 480 290 7 V1 (J·mol1·K1) 160 160 260 70 Annexe 3. Constantes d'acidité à 25 °C (R est une chaine alkyle). pKa(RCO2H/RCO2) 4 ; pKa(ROH/RO) 17 ; pKa(H3O+/H2O) = 0 ; pKa(H2O/HO) = 14 ; pKa(RH/R) 50 ; pKa(R2NH/R2N) 36 ; pKa(EtO2CCH2R/ EtO2CCHR) 25. Annexe 4. Constantes physicochimiques et autres. Masses molaires (g·mol1) : C = 12 ; O = 16 ; H = 1 ; Si = 28. Numéro atomique : Z(Cl) = 17 Densité de l'éthanol : d = 0,8. Rayons covalents : r(O) = 66 pm ; r(H) = 31 pm. Page 14/15 Annexe 5. RMN 1H (gamme de déplacements chimiques). 7 Proton H H d (ppm) Proton d (ppm) 0,81,3 H 3,44,2 1,52,4 O O 3,95,2 R 1,93,1 O H O 4,57,2 H H R H OH 1,92,8 H 2,32,9 6,59,0 H O 9,010,5 O R 2,33,7 O O R N H 3,34,2 O 610 O R N H H R' 1012 O R OH H Annexe 6. Constantes de couplage 1H1H usuelles.7 Protons H H H H H H H J (Hz) 68 812 1318 H H 03 7 H H H H 610 14 H 01 E. Prestsch, P. Bühlmann, M. Badertscher, Structure Determination of Organic Compounds, Springer-Verlag, Berlin, Heidelberg, 4 edn. 2009. Page 15/15