
Mines Chimie PC 2020
| Thème de l'épreuve | Synthèse totale de la fusarisétine A.. Le cobalt aux degrés d'oxydation +II et +III. |
| Principaux outils utilisés | chimie organique, orbitales moléculaires, chimie de coordination, solutions aqueuses, diagramme E-pH, cristallographie, cinétique chimique |
| Mots clefs | fusarisétine, fusarisétine A, complexe allyle, allylure, Tsuji-Trost, cobalt, oxydation, mécanisme inorganique, \Co3O4, spinelle, diffusion |
Corrigé
:👈 gratuite pour tous les corrigés si tu crées un compte
👈 l'accès aux indications de tous les corrigés ne coûte que 1 € ⬅ clique ici
👈 gratuite pour tous les corrigés si tu crées un compte
- - - - - - - - - - - - - - - - -
👈 gratuite pour ce corrigé si tu crées un compte
- - - - - - - - - - - - - - - - - - - - - - -
Énoncé complet
(télécharger le PDF)















Rapport du jury
(télécharger le PDF)





Énoncé obtenu par reconnaissance optique des caractères
A2020 --- CHIMIE PC
Cm
Concours commun
Mines-Ponts
ÉCOLE DES PONTS PARISTECH,
ISAE-SUPAERO, ENSTA PARIS,
TÉLÉCOM PARIS, MINES PARISTECH,
MINES SAINT-ÉTIENNE, MINES NANCY,
IMT ATLANTIQUE, ENSAE PARIS, CHIMIE PARISTECH.
Concours Centrale-Supélec (Cycle International),
Concours Mines-Télécom, Concours Commun TPE/EIVP.
CONCOURS 2020
ÉPREUVE DE CHIMIE
Durée de l'épreuve : 4 heures
L'usage de la calculatrice et de tout dispositif électronique est interdit.
Les candidats sont priés de mentionner de façon apparente
sur la première page de la copie :
CHIMIE - PC
L'énoncé de cette épreuve comporte 22 pages de texte.
Si, au cours de l'épreuve, un candidat repère ce qui lui semble être une erreur
d'énontcé, il le
signale sur sa copie et poursuit sa composition en expliquant les raisons des
initiatives qu'il est
amené à prendre.
Les sujets sont la propriété du GIP CCMEP. Ils sont publiés les termes de la
licence
Creative Commons Attribution - Pas d'Utilisation Commerciale - Pas de
Modification 3.0 France.
Tout autre usage est soumis à une autorisation préalable du Concours commun
Mines Ponts.
À 2020 chimie PC
DEBUT DE L'ENONCE
Des données utiles à la résolution du problème sont fournies à la fin de
l'énoncé.
Cette épreuve est constituée de deux parties indépendantes.
Synthèse totale de la fusarisétine À
En 2011, l'équipe du professeur Ahn a isolé un composé naturel, la fusarisétine
À, à partir
d'un champignon terricole, le Fusarium sp. FN080326. La fusarisétine À a
suscité l'intérêt
pour ses propriétés anticancéreuses, sans être cytotoxique.
Ce composé a une structure complexe ; il contient en particulier une jonction
Spiranique,
c'est-à-dire que deux de ses cycles sont reliés uniquement par un seul atome
commun
(figure 1). La stéréochimie de plusieurs des centres stéréogènes de la
fusarisétine À a été
intentionnellement omise.
Figure I - Structure de la (-)-fusarisétine À.
Ce problème est consacré à l'étude d'une partie de la synthèse totale de cette
molécule. Il est
composé de trois parties indépendantes.
Stratégie de synthèse
On étudie dans un premier temps la synthèse de la molécule À réalisée à partir
du (S)-(-)-
citronellal dont la structure est proposée ci-dessous. La synthèse de À publiée
par Inoue et
coll. se déroule en cinq étapes.
gs OTBS
a--ÿ LE TES : 3
Ù oO |
Une partie des conditions opératoires utilisées pour réaliser deux des étapes
de la
transformation du citronellal en aldéhyde À sont rassemblées ci-après :
Page 1 /22
Chimie 2020 Filière PC
conditions 1 conditions 2
a-4 EUR ho 708
nie es
I
CH»Ch Toluène
S Dean-Stark
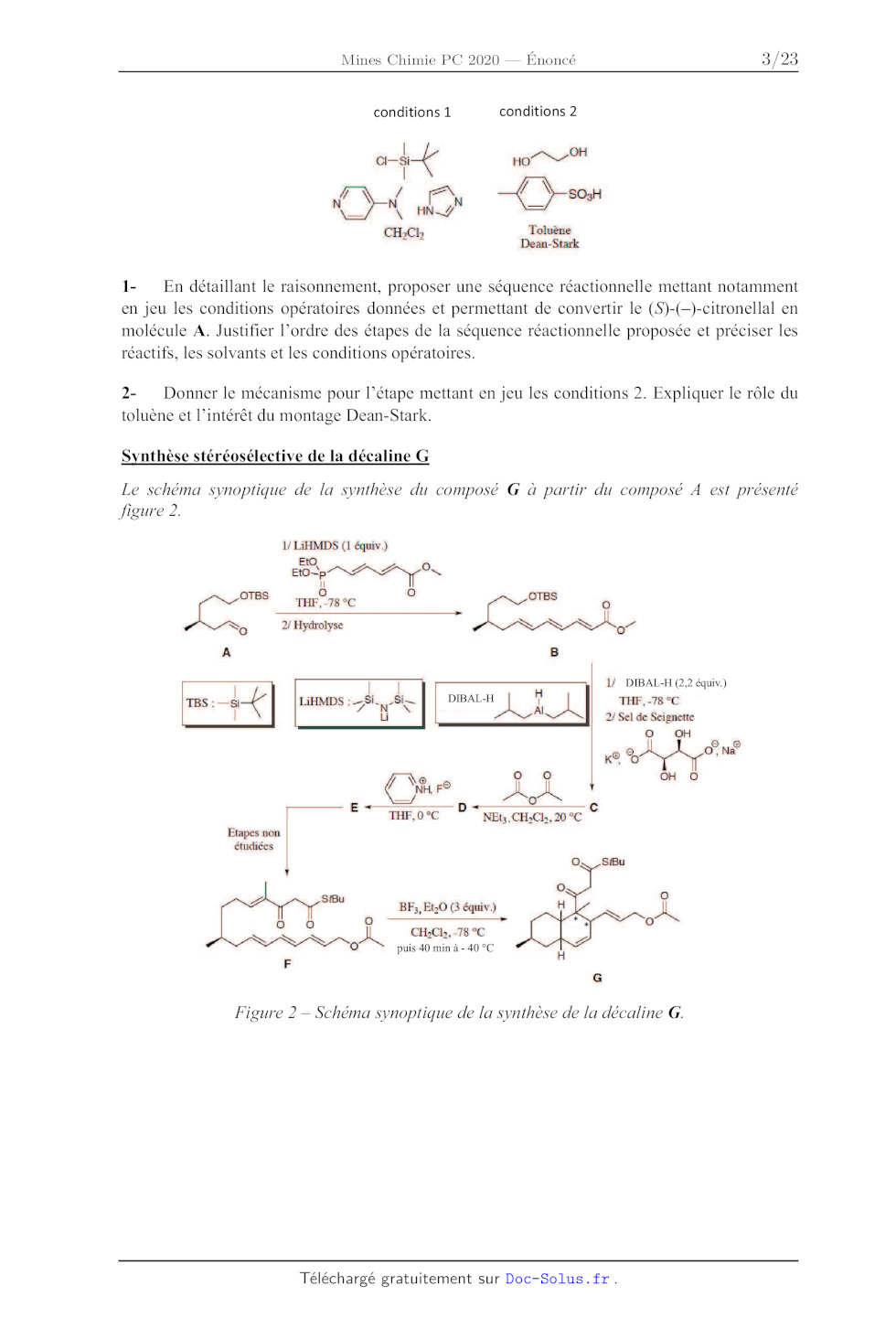
1- En détaillant le raisonnement, proposer une séquence réactionnelle mettant
notamment
en Jeu les conditions opératoires données et permettant de convertir le
(S)-(--)-citronellal en
molécule A. Justifier l'ordre des étapes de la séquence réactionnelle proposée
et préciser les
réactifs, les solvants et les conditions opératoires.
2- Donner le mécanisme pour l'étape mettant en jeu les conditions 2. Expliquer
le rôle du
toluène et l'intérêt du montage Dean-Stark.
Synthèse stéréosélective de la décaline G
Le schéma synoptique de la synthèse du composé G à partir du composé À est
présenté
figure 2.
1/ LiHMDS (1 équiv.)
E0-- POSTS
OTBS THE? me 0 OTBS ü
Lx 2/ Hydrolyse " Lan 0"
A B
1/ DIBAL-H (2,2 équiv.)
L.
TBS : a LiHMDS : Se | dat PT ke à fs À THE. -78 °C
L ds 2/ Sel de Seignette
O CH 5
@
O ,Na
9 © OH O
/ N9,.6
NH, F
é (} lle Pig c
THF,0 °C NEt: ,CH;Ch, 20 °C
Etapes non
étudiées
O4 _StBu
, O
SB
g ° BF;, Et:0 (3 équiv.) H . À
0. À: CH;Cb, -78 °C
O puis 40 min à - 40 °C H
F
G
Figure 2 -- Schéma synoptique de la synthèse de la décaline G.
Page 2 /22
Chimie 2020 Filière PC
Le composé À, énantiomériquement pur, est ajouté, à --78 °C, à une solution
contenant 1,0
équivalent molaire de LiHMDS (bis(triméthylsilyl)amidure de lithium) et un
léger excès de
phosphonate (composé phosphoré). Le milieu est hydrolysé et on isole, après
purification, le
composé B avec un rendement de 71 %.
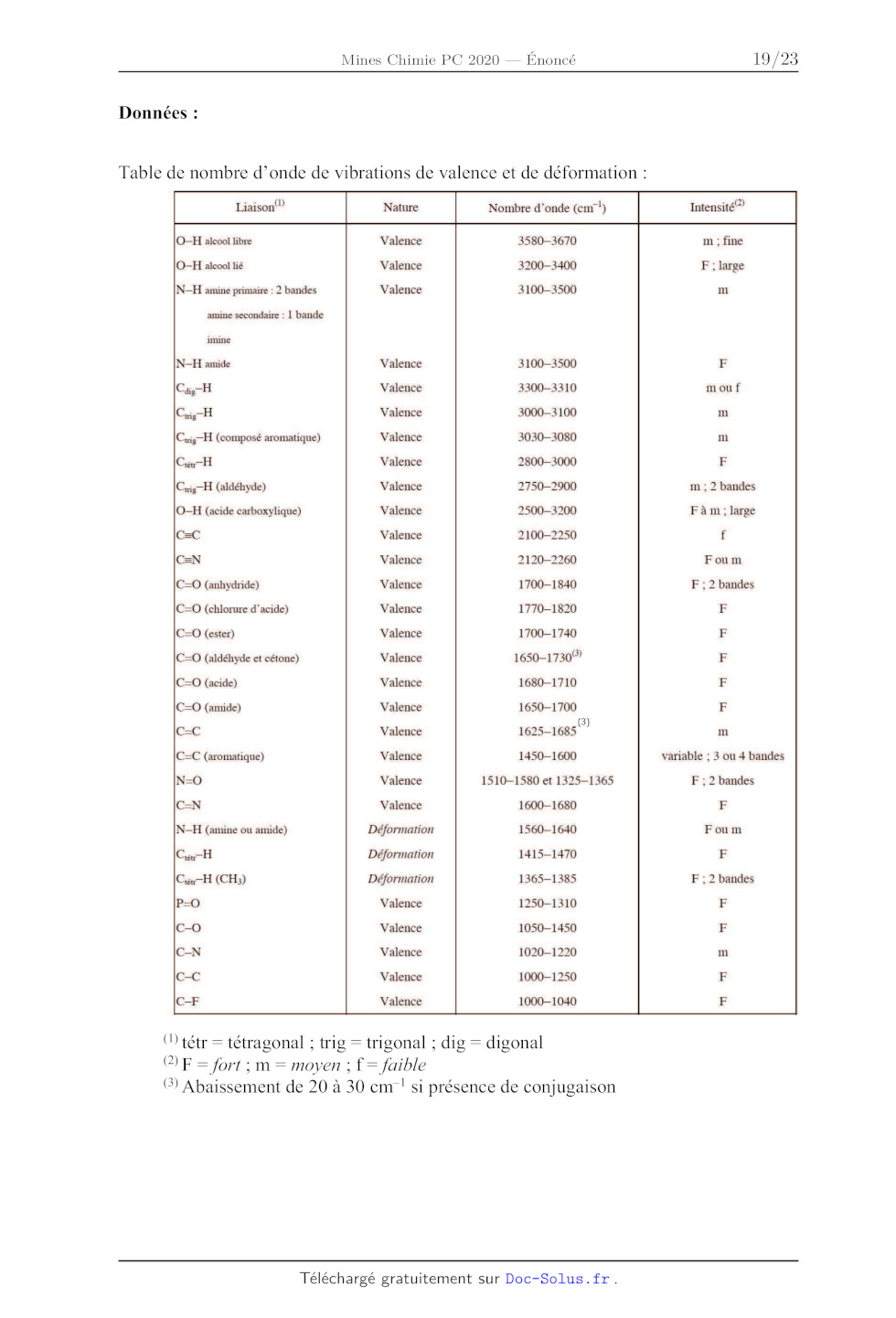
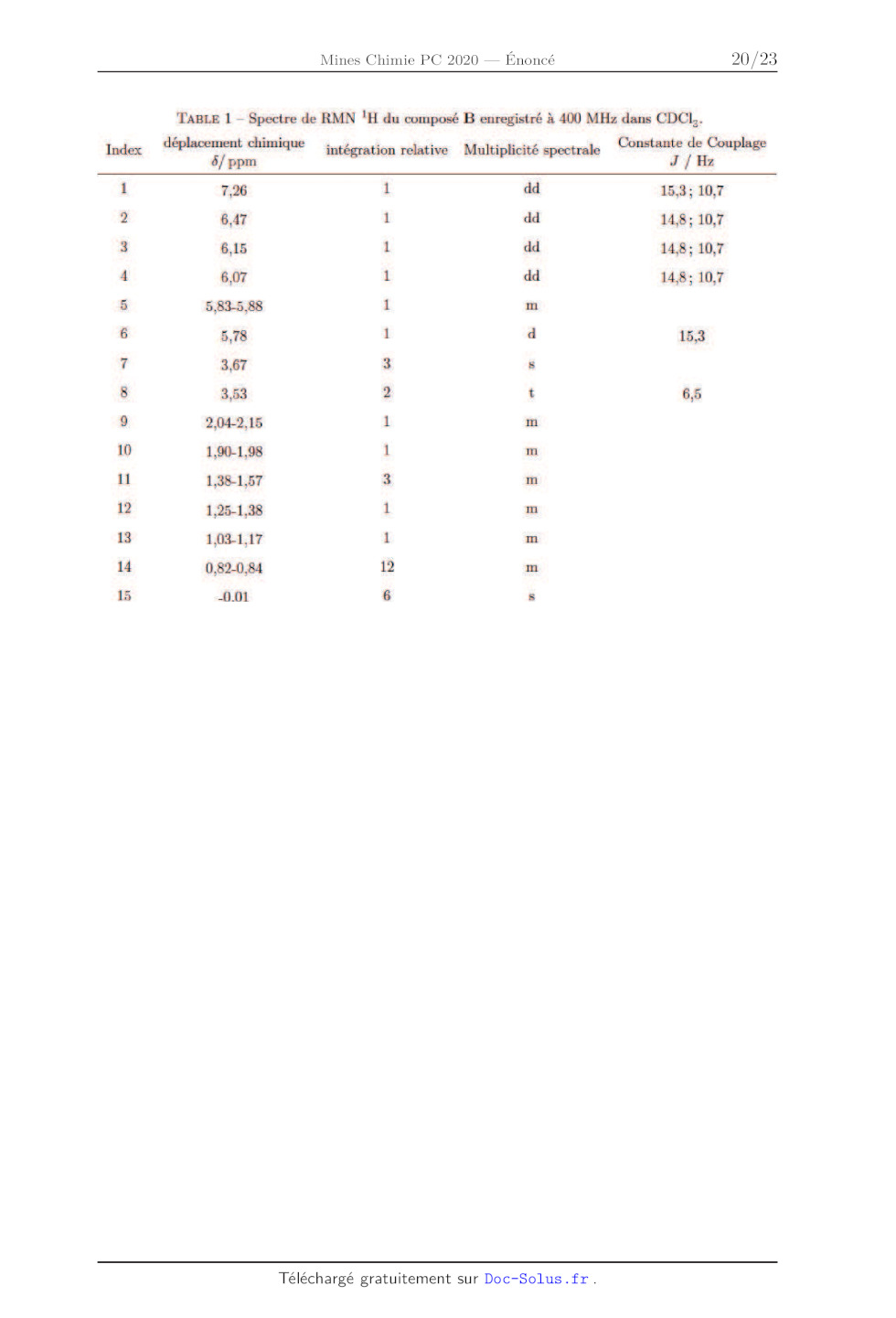
Le spectre de RMN !H du composé B, enregistré dans CDCI: à la fréquence de 400
MHz, est
présenté dans la table 1 donnée en annexe.
3- À quelle réaction classique s'apparente cette transformation ? Combien de
stéréo-
isomères sont susceptibles d'être obtenus ? Quelle est la relation de
stéréo-isomérie qui les
lie ?
4- En explicitant le raisonnement, attribuer les signaux n°* 1, 5, 6, 7 et 8
(cf. table 1) aux
protons qui en sont responsables.
En solution dans le THF, le composé B est traité, à --78 °C, par 2,2
équivalents molaires
d'hydrure de diisobutylaluminium en solution dans l'hexane. Le milieu est agité
à cette
température pendant 30 min, puis une solution saturée de sel de Seignette (cf.
figure 2) y est
ajoutée. La suspension obtenue est alors agitée vigoureusement jusqu'à
disparition complète
du précipité. Le contenu de la phase aqueuse est extrait par de l'éthanoate
d'éthyle, les
phases organiques sont rassemblées, séchées, le solvant est évaporé, puis le
produit EUR obtenu
est purifié par chromatographie. Lors de la transformation de B en EUR, le
spectre d'absorption
IR montre la disparition d'une bande à 1720 cm ! et l'apparition d'une bande à
3356 cm !.
5- Que peut-on déduire de la comparaison des spectres d'absorption IR de B et
de C ?
Donner la structure du produit C.
Le composé C est dissous dans du dichlorométhane contenant de la triéthylamine
en léger
excès. L'anhydride éthanoïque est ensuite ajouté goutte à goutte. Après deux
heures
d'agitation à température ambiante, une solution aqueuse saturée
d'hydrogénocarbonate de
sodium est ajoutée. Après traitement et purification, D est obtenu avec un
rendement de 96 %.
6- Donner la formule de D et proposer un mécanisme pour sa formation.
Finalement, D est solubilisé dans le THF dans un récipient en plastique. Une
solution
contenant un mélange d'acide fluorhydrique et de pyridine est ajoutée goutte à
goutte à 0 °C.
L'hydrolyse du milieu réactionnel est suivie de l'extraction de la phase
aqueuse par
l'éthanoate d'éthyle. Les phases organiques rassemblées sont lavées avec de la
saumure,
séchées, et le solvant est évaporé. E est obtenu, après purification, avec un
rendement de
93 %. Par rapport au spectre de RMN lH de la molécule D, on observe, entre
autres, la
disparition des signaux intégrant respectivement pour neuf et six protons, et
l'apparition d'un
singulet intégrant pour un proton, de déplacement chimique égal à 2,30 ppm.
7- Donner la structure du composé E. Analyser le rôle du groupe
tert-butyldiméthylsilyle
(TBS) dans la stratégie de synthèse adoptée par les auteurs.
Page 3 /22
Chimie 2020 Filière PC
E est transformé en F par une séquence réactionnelle non étudiée ici. Ce
composé est
converti, en présence de trifluoroborane, en solution dans l'éthoxyéthane, en
un seul composé
bicyclique G. Cette réaction intramoléculaire, stéréosélective, permet de créer
en une seule
étape quatre centres stéréogènes. Les stéréodescripteurs des centres
stéréogènes marqués
d'une étoile sont tous les deux (R).
8- Compléter la structure de la molécule G en indiquant la position (par
rapport au plan
moyen de G) des substituants présents sur les centres stéréogènes contrôlés au
cours de
l'étape de cyclisation. On justifiera soigneusement le raisonnement utilisé
pour expliquer les
positions proposées.
9- Quel peut être le rôle du trifluoroborane dans cette synthèse ?
Étude de l'étape clé de la synthèse : formation d'un complexe n°-allyle du
palladium
Dans leur analyse rétrosynthétique, les auteurs ont proposé de former le cycle
à cinq atomes
de carbone à l'aide d'une réaction de substitution allylique. Cette
transformation a été, en
pratique, réalisée en trois étapes à partir du composé G (figure 3). Elle
exploite la capacité
qu'ont les complexes de palladium au degré d'oxydation 0 à former des complexes
7°-allyle
possédant un ligand appauvri en électron (n° indique ici la coordination de
trois atomes
contigus du ligand). L'objet de cette partie est d'étudier les caractéristiques
de cette
transformation.
O StBu O O
O
: © à
K:CO; (2 équiv.)
ÿ 0 À L S ééi
MeOH, CH;:Cl:
H H
G H
ñ
mn à.
O
NEt: (5 équiv.)
CH;Cb, 0 °C
«
Pd(OAc); (0,5 équiv.)
PBu; (1,25 équiv.) a
CH;CN
Figure 3 -- Formation stéréosélective du cycle à cinq atomes de carbone à
l'aide d'une
réaction palladocatalysée.
Page 4 /22
Chimie 2020 Filière PC
L'ester G est traité par du carbonate de potassium dans un mélange de méthanol
et de
dichlorométhane pour conduire après hydrolyse au cétoester H. L'analyse RMN du
proton du
produit H dans le chloroforme deutéré à température ambiante indique qu'il
existe sous deux
formes dans ces conditions. En plus des pics attendus pour la forme majoritaire
(95 %) du
cétoester H, on relève dans le spectre RMN obtenu deux signaux supplémentaires,
un singulet
intégrant pour un proton à à égal à 12,3 ppm, et un singulet intégrant pour un
proton à à égal
à 5,12 ppm.
10- Comment s'appellent les deux formes dont 1l est question pour le cétoester
H ? Dessiner
la structure de la forme minoritaire du composé H, puis attribuer les protons
aux pics
supplémentaires observés dans le spectre RMN du proton.
Le cétoester H est solubilisé dans le dichlorométhane. À la solution obtenue
sont
successivement ajoutés de la triéthylamine et le chlorure de l'acide
méthanesulfonique. On
suppose qu'il se forme transitoirement dans le mélange réactionnel un composé,
noté IL, qui
réagit in situ pour conduire à l'ester tricyclique I
11- Quel composé est habituellement préparé par action du chlorure de l'acide
méthanesulfonique sur un alcool, et quel est son intérêt ? Proposer alors une
structure pour
l'intermédiaire réactionnel I1.
12- Proposer un mécanisme schématique pour la transformation de I1 en I.
Le cycle catalytique postulé pour la formation du cétoester J à partir de
l'ester I (figure 3) est
représenté figure 4.
Pd&OAc h
L
| O
' SSL
ASS Nu Y
PdL> O
Etape 4 Etape 1
£ Nu Z O
n" LT
.Pd PAG, O
L L L
Etape 3 Etape 2
©
Nu° x+
Figure 4 -- Cycle catalytique postulé pour la réaction de Tsuji-Trost.
Page 5 /22
13-
Chimie 2020 Filière PC
la figure 4, dans lequel L est un ligand neutre.
Donner, en justifiant le raisonnement, le nom des étapes 1, 2 et 4 du cycle
catalytique de
Le complexe 17-allyle du palladium X* est composé formellement d'un complexe de
palladium chargé positivement et d'un ligand allylure (prop-2-én-l-ure). Ce
complexe X°
présente une réactivité électrophile que nous allons essayer d'expliquer en
étudiant son
diagramme d'orbitales moléculaires (O.M.).
Le diagramme des O.M. du complexe X* est construit par interaction entre les
orbitales du
fragment PdL>* -- où l'angle (L-Pd-L) est égal à 90° -- et celles du fragment
allylure C3H..
On suppose que ces deux fragments appartiennent à des plans perpendiculaires
entre eux.
L'atome de palladium et les deux ligands L appartiennent au plan (xOz), tandis
que les trois
atomes de carbone numérotés 1 à 3 du fragment allylure appartiennent à un plan
parallèle au
plan (yOz). On suppose aussi que les atomes de carbone I et 3 du fragment
allylure
appartiennent au plan (xOz). Le diagramme d'interactions simplifié entre les
O.M. des deux
fragments est donné figure 5. Seules les O.M. pouvant interagir sont
représentées.
y > |©
. _ ]2® \
ee" Pd x 17
Z 3
LE
po. ?6
' TT ps Ta
mm, (5 ;
! ' ! "
1 ' ".
dy ' : j Ts,
4
dxy ..! g | Ti
CT ---- D
:
# ,
+
'
dr, F a
dy2 72 _ é
_
O.M. PdL5®
OM. CH
Figure 5 -- Diagramme simplifié d'O.M. du complexe X*. Les O.M. du complexe
PdL" sont
indiquées en vis à vis pour plus de clarté.
Page 6 /22
Docinment 1 Première série d'expériences
RC RO RENTE TP O Ne TAROT TNT NTI SON TETE
En
os TT DENA
Aucune 1éacuon
L CN
À AA
ECO
ESS
PRES
o
LCL
" NES NOTE AN A CETTE TT
ESS LUS
CRETE ECRETN
DEN TP PS CR LC NÉE
Chimie 2020 Filière PC
Document 2 --- Seconde série d'expériences
HaAYASHI et coll. * ont réalisé l'expérience suivante afin d'étudier la
sélectivité de la réaction de
TsUuI-TROST :
A/B : 92/8
Ss Ph Pd / dppe Ss Ph SL Ph
EUR es Lu. à LÉ
se Li CH(COOMe)) CH(COOMe)
0 eo A B
69,5 % (S) THF,20 h 68.5 % (S) 65 % (R)
30.5 % (R) 31.5 % (R) 35 %(S)
a. HayasHt et coll., J. Org. Chem., 1986, 51, 723 - 727.
J
17- Expliquer les résultats expérimentaux fournis au document 1 (où L = PPh3),
et conclure
quant à la stéréochimie de l'étape 2 (cf. figure 4). On pourra représenter dans
leur
conformation chaise les cyclohexanes substitués étudiés.
18- De la même manière, analyser les résultats du document 2 et discuter de la
sélectivité de
l'étape 3 (cf. figure 4) dans ce cas. Le dppe
(1,2-Bis(diphénylphosphino)éthane) est un ligand
bidentate.
19- En exploitant les résultats des questions précédentes, proposer le cycle
catalytique
permettant d'expliquer la formation de J à partir de I. Préciser, à chaque
étape, la
configuration des différents intermédiaires réactionnels.
cé
Page 8 /22
Chimie 2020 Filière PC
Le cobalt aux degrés d'oxydation +2 et +3
Le cobalt est l'élément de numéro atomique 27. Relativement peu abondant, il
est critique
pour de nombreuses applications en tant qu'élément de super-alliages,
d'aimants, de
céramiques, de matériaux pour les batteries ou encore de catalyseurs. Ce
problème
s'intéresse plus particulièrement aux réactions impliquant le cobalt aux degrés
d'oxydation
+2 et +3 en solution, ainsi qu'à l'oxyde Co304.
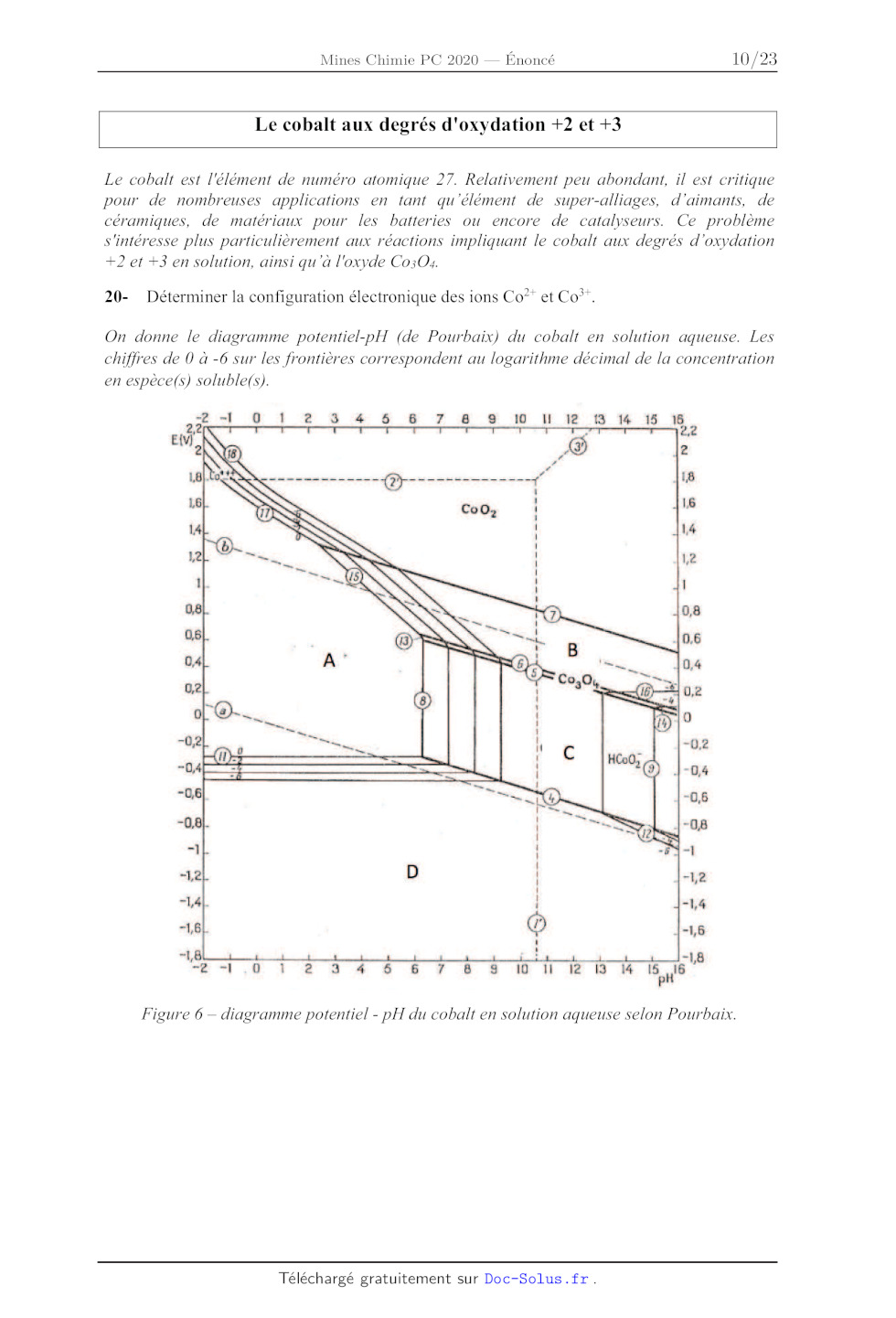
20- Déterminer la configuration électronique des ions Co?* et Co*'.
On donne le diagramme potentiel-pH (de Pourbaix) du cobalt en solution aqueuse.
Les
chiffres de 0 à -6 sur les frontières correspondent au logarithme décimal de la
concentration
en espèce(s) soluble(s).
13 14
F1 '
Lei
2 | Q 1 ?2 12 15 16
T T T 2,2
« -- 2,
2 © le
L8|.
L6
L4
J,2
0,6
0,4
0,2
se *
1,41.
1,6
-1,8
un
IL
0,81
l
@)
À
--
ni
4
/
L
/
=_-- = no me ne ce fe de dt
Si)
=
f
/
SSe Éd
/
L
|
béméhnes
]
0 ee 3 4
Ô
6
7
B
3
L, 2.8 i i L i
10 11 12 13 5 ;
118
116
?
16
ph
Figure 6 -- diagramme potentiel - pH du cobalt en solution aqueuse selon
Pourbaix.
Page 9 /22
Chimie 2020 Filière PC
21- Affecter aux domaines A, B, C et D les espèces chimiques correspondantes. A
quoi
correspondent les lignes en pointillés (a) et (b) dans le diagramme ? Retrouver
par un calcul la
pente de la droite (15) séparant les domaines A et B.
Les ions Co°*x en solutions aqueuses précipitent facilement en milieu basique
pour former de
l'hydroxyde de cobalt, utilisé notamment comme matériau (ou précurseur de
matériaux) pour
les batteries.
22- Déterminer par le calcul la concentration maximale en ion Co?':4 dans une
solution de
soude diluée à pH--9,5, et confronter ce résultat au diagramme potentiel-bH.
Montrer qu'il est
en revanche possible d'obtenir une solution de Co(Il) de concentration c=0,01
mol.L:'! à
pH=9,5 en milieu ammoniacal. On considérera que le seul complexe formé avec
l'ammoniac
est Co(NH:)6°".
23- Calculer le potentiel standard du couple Co(NH:)6**/Co(NH:)6*. Comparer la
stabilité
de l'ion Co** en milieu ammoniacal par rapport à sa stabilité en milieu aqueux,
du point de
vue oxydoréduction.
Dans un article fondateur publié en 1951 sur les cinétiques et mécanismes de
substitution
dans les complexes en solution, Henry Taube (prix Nobel de chimie en 1983)
écrit :
"En utilisant les données rapportées par J. Bjerrum, on peut montrer qu'à 25 °C
dans les
conditions [H']=1 mol.L'} et [NH4']=1 mol.L'|, la réaction
Co(NH3)e*+ H* + H20 = Co(NH:):(H20)* + NH,
est quasiment totale à l'équilibre, et seulement 0,01 % environ des ions
Co(NH:)6" restent
inchangés. La force motrice pour une substitution plus poussée de l'ammoniac
est encore plus
grande. Malgré cette très grande force motrice favorisant la conversion en
complexe aquo en
milieu acide, la réaction est incommensurablement lente. Si on considère à
présent la
réaction en milieu basique
Co(NH3)6*+ 3 HOT = Co(OH)3s +6 NH,
et qu'on utilise, en addition aux données de Bjerrum pour la stabilité de
Co(NH:)6*, la
valeur donnée par Latimer pour le produit de solubilité de Co(OH);., on calcule
une
concentration [Co(NH:)6*]-10" mol.L! à l'équilibre avec Co(OH);s quand [OH ']-1
mol.L'! et [NH;]=1 mol.L'!. La réaction reste lente, quoique plus rapide qu'en
milieu acide."
24- Vérifier les deux calculs de H. Taube à l'aide des données en annexe. Que
montrent-
ils ?
Page 10 /22
Chimie 2020 Filière PC
On peut facilement précipiter des sels de CoClB:(NK)5, qui se dissolvent en
solution aqueuse
en formant le complexe CoCI(NH:)5 ** dont la réactivité fait l'objet des
questions suivantes.
On s 'intéressera plus particulièrement à deux réactions : la substitution de
CT" et l'oxydation
du Cr).
La substitution de CT par HO est lente (du moins à l'abri de la lumière), ce
qui facilite la
mise en oeuvre expérimentale d'un suivi cinétique. Elle a de fait été très
étudiée afin d'en
élucider le mécanisme.
25- En supposant un mécanisme de type Sx2, donner la loi cinétique attendue, en
notant ko
la constante cinétique.
En 1966, S.C. Chan réalisa des mesures cinétiques en solutions avec un large
excès d'ions
HO et pour des degrés d'avancement faibles en raison de la lenteur de la
substitution. 11
détermina la constante cinétique de pseudo ordre un, notée ko», et définie par
kobs=ko[HO ],
pour différentes concentrations en ions HO. Il traça alors le graphe de la
figure 7
représentant l'évolution de la constante kr, en fonction de la concentration en
ions HO :
10°, (sec!)
© ml os
Q OO4 O'OB
(on
Figure 7 -- Evolution de kors, Constante de vitesse de pseudo ordre un de la
réaction
d'hydrolyse de CoCI(NH:);", en fonction de [HO] à 25 °C.
Page 11 /22
Chimie 2020 Filière PC
26- Expliquer pourquoi ce tracé remet en cause l'hypothèse d'un mécanisme Sx2.
Afin d'interpréter ses résultats, S.C. Chan proposa le mécanisme suivant,
baptisé Sn2 IP
(IP=ion pair) :
K
CoCI(NH.).2* + OH: = {CoCI(NH.).2* OH}
équilibre paire d'ions
rapide
{COCI(NH;).2* OH} +, {Co(OH)(NH,).2* CI }
étape lente
{Co(OH)(NH;),2* CE} -- > Co(OH)(NH,).** + CF
rapide
On notera a la concentration initiale en complexe de cobalt et x la
concentration à l'équilibre
de la paire d'ions.
27- Exprimer x en fonction de K, a et [HO |.
28- Montrer qu'on peut écrire l'égalité kx=ktsa et en déduire l'expression de
la constante
cinétique du pseudo ordre un kit, en fonction de K, k et [HO |.
S.C. Chan traça alors la courbe de la figure 8 :
80+-
1/ #, bs (52EUR)
1
O 40
20
/loH]
Figure 8 -- Evolution de l'inverse de la constante cinétique de pseudo ordre un
de la réaction
d'hydrolyse de CoCI(NH:); * en fonction de l'inverse de la concentration [HO'],
à 25 °C.
Page 12 /22
Chimie 2020 Filière PC
29- Ce résultat est-1l en accord avec le modèle cinétique Sx2 IP ? Expliquer
comment
déterminer les paramètres K et k du modèle.
On considère à présent le mécanisme suivant de type SxI1 CB (CB=conjugate base)
:
K'
CoCI(NH.).2* + OH = CoCI(NH.),(NH) * + H,0
équilibre
rapide
K 2+ -
CoCI(NH;),(NH) * ---- Co(NH;),(NH,) #* + CI
étape lente
Co(NH;),(NH;,)%* +40 --> Co(OH)(NH;).7*
rapide
30- Montrer que les résultats de l'étude cinétique précédente ne permettent pas
de
discriminer entre le mécanisme Sx2 IP proposé par S.C. Chan et un mécanisme Sxi
CB.
Une étude de fractionnement isotopique de l'oxygène a été menée afin de
trancher entre les
deux mécanismes.
[60|/[180] dans le complexe hydroxo
[160]/[180] dans le solvant (eau)
On appelle fractionnement isotopique le rapport f --
Le complexe hydroxo étant ici Co(OH)(NH3)5 *. Chaque entité (ion complexe
hydroxo ou
molécule d'eau) comportant un (seul) atome d'oxygène, le rapport ['$0]/[*O] est
égal à la
concentration de cette entité où l'oxygène est sous forme 160 divisée par la
concentration de
cette même entité où l'oxygène est sous forme 0.
On indique que la réaction d'équation H2'*O + OH = H2%0O + OH a pour constante
d'équilibre :
Kech=1,040 (+/-0,003) à 25 °C.
Le fractionnement isotopique mesuré pour Co(OH) (NH): ** a pour valeur :
f=1,005 (+/- 0,001).
31- Parmi les deux mécanismes potentiels de la réaction de substitution, Sx2 IP
ou Sxi CB,
quel est celui qui rend le mieux compte de ce résultat ? Proposer une
explication au fait de ne
pas avoir obtenu pour fractionnement isotopique f-1,000.
Page 13 /22
Chimie 2020 Filière PC
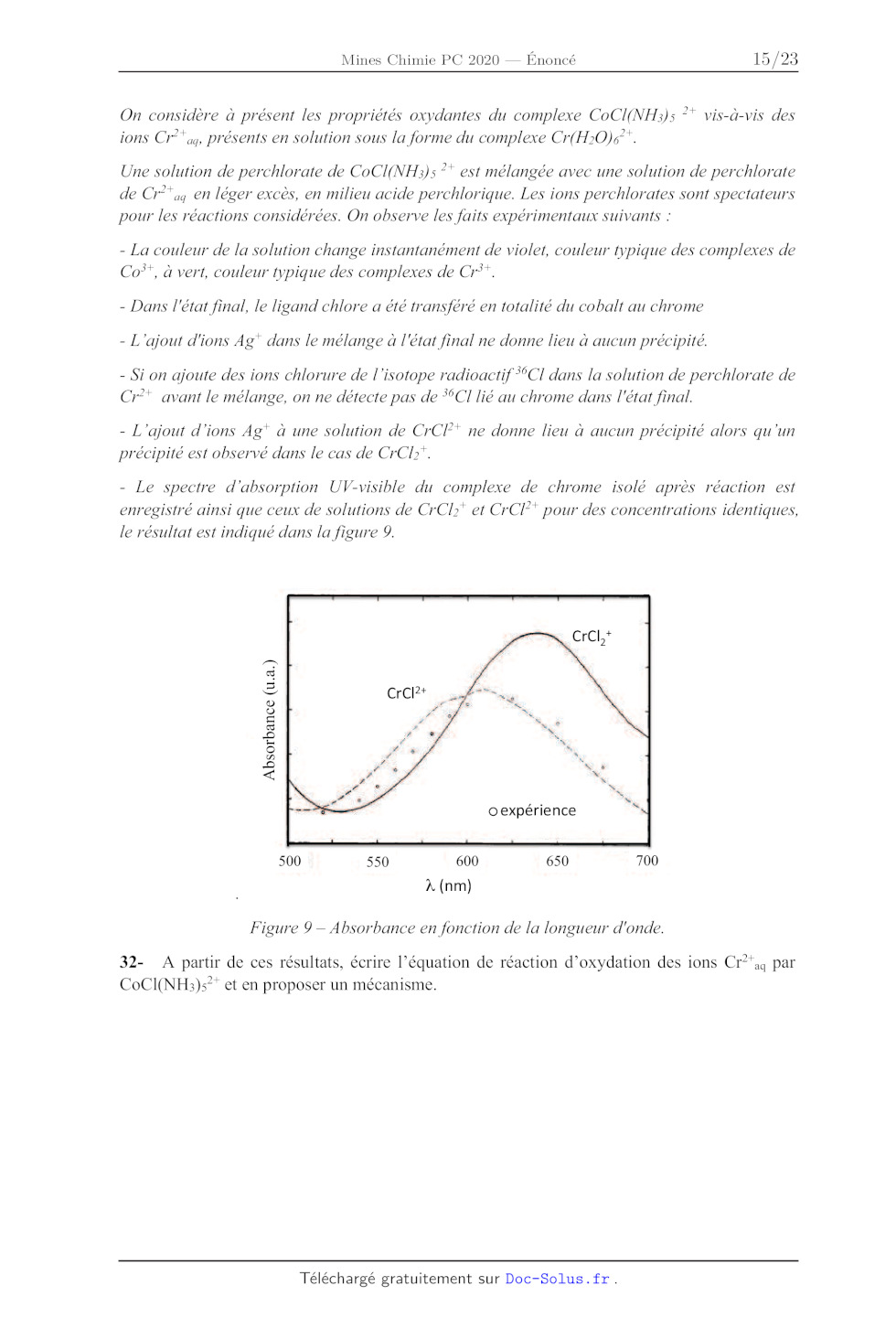
On considère à présent les propriétés oxydantes du complexe CoCl(NH;)5 ?*
vis-à-vis des
ions Cr°* ay présents en solution sous la forme du complexe Cr(H>0)é*.
Une solution de perchlorate de CoCI(NH;3): ** est mélangée avec une solution de
perchlorate
de Cr°"ay en léger excès, en milieu acide perchlorique. Les ions perchlorates
sont spectateurs
pour les réactions considérées. On observe les faits expérimentaux suivants :
- La couleur de la solution change instantanément de violet, couleur typique
des complexes de
Co**, à vert, couleur typique des complexes de Cr'*.
- Dans l'état final, le ligand chlore a été transféré en totalité du cobalt au
chrome
- L'ajout d'ions Ag" dans le mélange à l'état final ne donne lieu à aucun
précipité.
- Si on ajoute des ions chlorure de l'isotope radioactif *°CI dans la solution
de perchlorate de
Cr°* avant le mélange, on ne détecte pas de "(CI lié au chrome dans l'état
final.
- L'ajout d'ions Ag* à une solution de CrCl* ne donne lieu à aucun précipité
alors qu'un
précipité est observé dans le cas de CrCl".
- Le spectre d'absorption UV-visible du complexe de chrome isolé après réaction
est
enregistré ainsi que ceux de solutions de CrCl:* et CrCF* pour des
concentrations identiques,
le résultat est indiqué dans la figure 9.
Absorbance (u.a.)
oexpérience
500 550 600 650 700
À (nm)
Figure 9 -- Absorbance en fonction de la longueur d'onde.
32- A partir de ces résultats, écrire l'équation de réaction d'oxydation des
ions Cr?'aq par
CoCI(NH)s"* et en proposer un mécanisme.
Page 14 /22
Chimie 2020 Filière PC
La partie suivante est consacrée à l'oxyde Co30} et à certaines de ses
propriétés.
La structure cristallographique de Co3O1 est celle d'un spinelle. Les ions
oxygène forment un
empilement cubique à faces centrées, les ions Co°* occupent des sites
tétraédriques et les ions
Co** des sites octaédriques. La structure est représentée dans la figure 10 en
coupant la
maille en deux dans le sens frontal pour faciliter son analyse. Le paramètre de
maille vaut
806,4 pm.
OO" eCo* o Co"
Figure 10 -- Structure cristallographique du Co30O4.
33- Quelles sont les fractions des sites tétraédriques et octaédriques occupées
dans cette
structure ?
34- Retrouver par le calcul la masse volumique de Co304 indiquée dans les
données en
annexe.
35- Calculer le rayon d'un site tétraédrique et le comparer au rayon ionique de
Co'*.
Commenter.
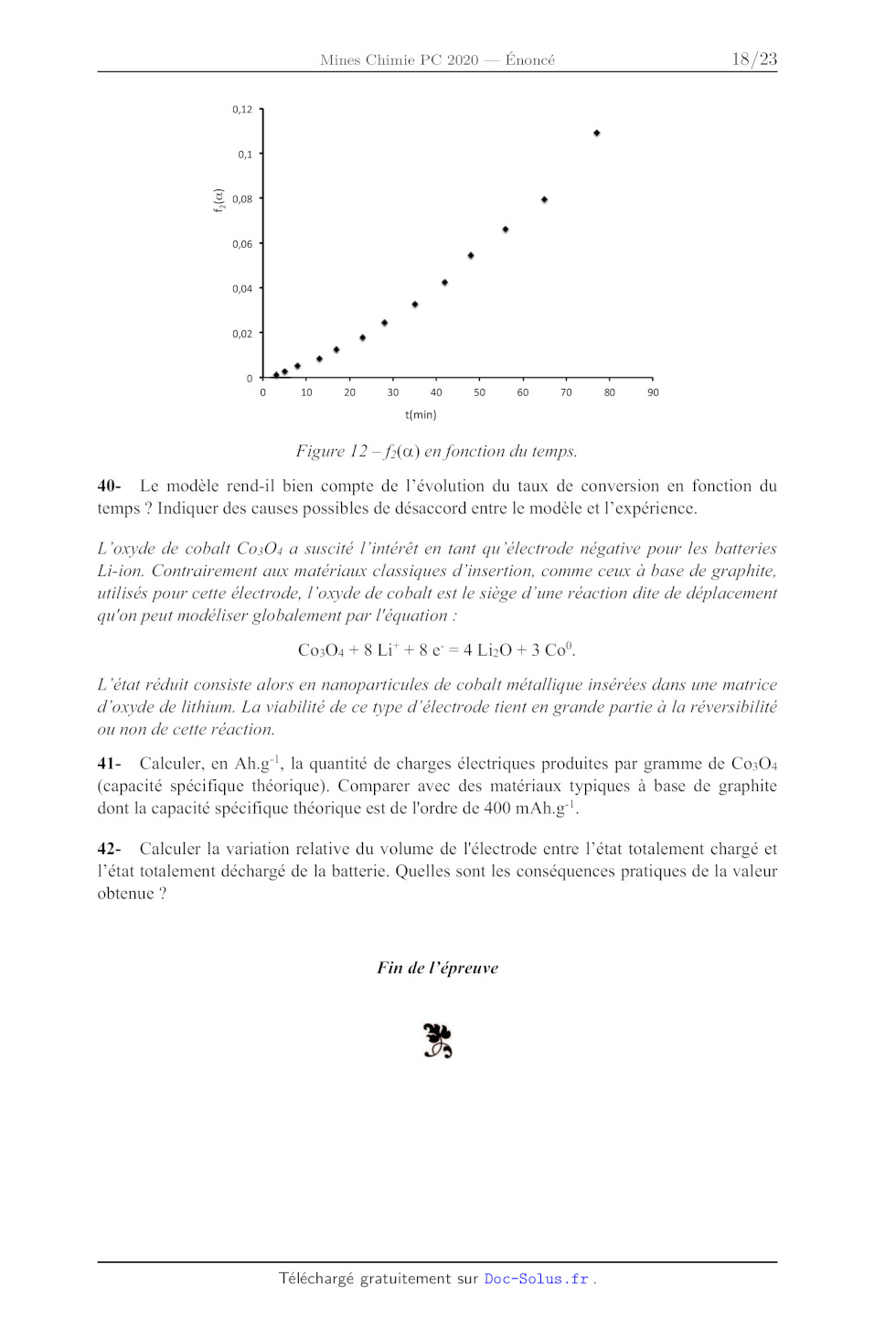
On étudie à présent la transformation de Co3O:4 en CoO par traitement thermique.
36- En faisant une ou plusieurs approximation(s) qu'on justifiera, évaluer à
l'aide des
données la température de décomposition de Co304 en CoO sous air.
Cette transformation se propage en fait depuis la surface des cristaux de Co3O4
jusqu'à leur
coeur. On considère des particules sphériques de rayons ro à t-0 et une loi de
type r(t)=ro-kt
avec r(t) le rayon du coeur de Co304 à la date t.
37- Exprimer la loi fi(o«)=kt reliant la fraction à de Co304 converti en
fonction du temps t.
Page 15 /22
Chimie 2020 Filière PC
En réalité, la diffusion de l'oxygène à travers la couche de CoO qui se forme
autour de Co304
est limitante. On considère, pour simplifier, que les interfaces sont planes
comme indiqué
dans la figure 11. On note D le coefficient de diffusion du dioxygène, C; sa
concentration à
l'interface CoO/Co:304 supposée constante, et Co sa concentration en surface
supposée
constante également.
CoO Co;,0,
EURO 4 + 002EUR <- 009 X dx Figure 11 -- Schéma de diffusion à travers une couche de CoO. 38- Exprimer le flux sortant en dioxygène à partir de la loi de Fick. En écrivant l'égalité entre la quantité de dioxygène créée par la réaction à l'interface et celle diffusée à travers la couche de CoO, en déduire qu'on a une loi de type x?=k't avec x épaisseur de la couche de CoO (il n'est pas demandé d'expliciter k'). 39- En supposant que l'épaisseur de CoO créée est égale à l'épaisseur de Co304 consommée et que la loi établie est transposable à des particules sphériques de rayon ro, en déduire la nouvelle expression f(a)-k't de la relation entre @ et t. Une expérience faite à 1060°C donne les résultats indiqués dans le figure 12 : Page 16 /22 Chimie 2020 Filière PC 0,1 - Y 0,08 - + 0,06 " 0,04 - 0,02 - . 0 10 20 30 40 50 60 70 80 90 t(min) Figure 12 -- fa) en fonction du temps. 40- Le modèle rend-1l bien compte de l'évolution du taux de conversion en fonction du temps ? Indiquer des causes possibles de désaccord entre le modèle et l'expérience. L'oxyde de cobalt Co3O:4 a suscité l'intérêt en tant qu'électrode négative pour les batteries Li-ion. Contrairement aux matériaux classiques d'insertion, comme ceux à base de graphite, utilisés pour cette électrode, l'oxyde de cobalt est le siège d'une réaction dite de déplacement qu'on peut modéliser globalement par l'équation : Co3O4 + 8 Li' +8 e° = 4 LibO + 3 Co. L'état réduit consiste alors en nanoparticules de cobalt métallique insérées dans une matrice d'oxyde de lithium. La viabilité de ce type d''électrode tient en grande partie à la réversibilité ou non de cette réaction. 41- Calculer, en Ah.£g'!, la quantité de charges électriques produites par gramme de Co30O4 (capacité spécifique théorique). Comparer avec des matériaux typiques à base de graphite dont la capacité spécifique théorique est de l'ordre de 400 mAh.g"!. 42- Calculer la variation relative du volume de l'électrode entre l'état totalement chargé et l'état totalement déchargé de la batterie. Quelles sont les conséquences pratiques de la valeur obtenue ? Fin de l'épreuve F Page 17 /22 Chimie 2020 Filière PC Données : Table de nombre d'onde de vibrations de valence et de déformation : Liaison(? Nature Nombre d'onde (enr !) Intensité? O-H alcool libre Valence 3580-3670 m ; fine O-H alcool lié Valence 3200-3400 F ; large NH amine primaire : 2 bandes Valence 3100-3500 m amine secondaire : 1 bande imine NH amide Valence 3100-3500 F Cäig--H Valence 3300-3310 m ou f Cuig-H Valence 3000-3100 m Crig--H (composé aromatique) Valence 3030-3080 m Crée H Valence 2800-3000 F Crig--H (aldéhyde) Valence 2750-2900 m ; 2 bandes O-H (acide carboxylique) Valence 2500-3200 F à m ; large Cac Valence 2100-2250 f C=N Valence 2120-2260 F ou m C=0O (anhydride) Valence 1700-1840 F ; 2 bandes C=0O (chlorure d'acide) Valence 1770-1820 F C=0O (ester) Valence 1700-1740 F C=O (aldéhyde et cétone) Valence 1650-1730 F C=0O (acide) Valence 1680-1710 F C=0O (amide) Valence 1650-1700 F CL Valence 1625-1685 m C=C (aromatique) Valence 1450-1600 variable : 3 ou 4 bandes N=0 Valence 1510-1580 et 1325-1365 F ; 2 bandes C=N Valence 1600-1680 F NH (amine ou amide) Déformation 1560-1640 F ou m CiéxH Déformation 1415-1470 F Cia-H (CH) Déformation 1365-1385 F : 2 bandes P-O Valence 1250-1310 F C-O Valence 1050-1450 F C-N Valence 1020-1220 m CC Valence 1000-1250 F C-F Valence 1000-1040 F (D tétr = tétragonal ; trig = trigonal ; dig = digonal @F = fort ; m = moyen ; Î = faible 6) Abaissement de 20 à 30 cm ! si présence de conjugaison Page 18 /22 TABLE 1 Spoetre de RMN TH du composé B enregistré rent RUB IP SRE RER ANT Bride Dee intégration relative Multiplicité spectrale 1 L To l ou REIN 2 Cens il dd FEI El re ll dd EEE ll CA il dd EST nl HE 1 [ E EE L Es In 1 UT J ë à L ll Le il RTE ll ui ol RES il m ni RS 5 nm nl 125-1.3S il on E HSE l nm 11 DST el M ns AT n 5 Chimie 2020 Filière PC Groupe fonctionnel Déplacement chimique Structure Jan Typical range value Hg--c Li si CH;--CH;-- 6-8 7 = c--CH,--c-- L CH;--CH< 5-7 5 H£ -- ( --0-- --|- -- C=CH -- -- CH ;-- CH 2 5-8 7 H3C -- C -- -- I M # HC--NT --_-- soeur _ > C=CH--CHC #11 6
R-- CH, --C -- -- w, #
! /C=CH---CH=C 6-13 11$
H3C-- 0 -- +
R-- CH, --0-- 7 YCH--CHO 0-3 2
R-- OH
>e=cm T ... YC=CH--CHO 58 7
-- CH = CH--
ex" à cis-CH--CH-- 012 8
"EN :
trans-CH=CH-- 12-18 15
R L =0 +
n--f--04 Constantes de couplage entre protons portés par des
0 à L S À L + carbones vicinaux (en Hertz)
ë (ppm
CR nes (source: Constantes des spectres RMN par Nicole
Platzer -- banque documentaire « Techniques de
déplacements chimiques en fonction de la nature | l'Ingénieur »)
chimique du groupement fonctionnel (en ppm par
rapport au TMS)
Constantes d'équilibre acido-basiques :
Couple pK'A
eg
Ion pyridinium / pyridine 5
Et,NH* / NEt; 12
ROH / RO 17-19
(Valeurs données dans l'eau, à T = 298 K ; R est un groupe alkyle)
Page 20 /22
Chimie 2020 Filière PC
Constante d'Avogadro : Na= 6,0.107* mol'!
Constante des gaz parfaits : R = 8,3 J.K'!.mol'!
RT
Constante de Nernst à 298 K : TR m0 = 0,06V
Constante de Faraday : 96500 C.mol!
Produit ionique de l'eau à 298 K : Ke = 10°!*
Potentiels standard par rapport à l'électrode standard à hydrogène à 298 K et
pH = 0:
ECo*'/Co°*) = 1,77 V
E(Cr**/Cr?*) =-0,41 V
E°(L1 /Li) = -3,04 V
Constantes de solubilité à 298 K :
Co(OHh2, = Co?" +2 HO pK; = 15
Co(OH}3, = Co*' +3 HO° pK; = 42
AgCI, = Ag* + CT pK; = 10
Constantes de dissociation des complexes à 298 K :
Co(NH:)6* = Co +6 NH3 pKp amm3 = 35
Co(NH:})6" = Co'* +6 NH3 pKp amm.2 = 5
Constante acido-basique à 298 K :
NH4° + H20 = NB + H30° pK: = 9,2
Constante de complexation à 298 K :
Co(H20)(NB3)5* + NH = Co(NBB)6* + H20 pKç -- -4,4
Masses molaires atomiques :
M(Co) = 59 g.mol!, M(O)-16 g.mol'!, M(Li) = 7 g.mol'!
Rayons ioniques :
rco_ (en environnement tétraédrique) = 56 pm; rco°' =
69 pm; ro? = 126 pm
Masses volumiques :
p(Co301) = 6,1 g.cm"
p(Co) = 8,9 g.cm"
p(CoO) = 6,4 g.cm"*
p(LiO) = 2,0 g.cm"
Page 21 /22
Chimie 2020 Filière PC
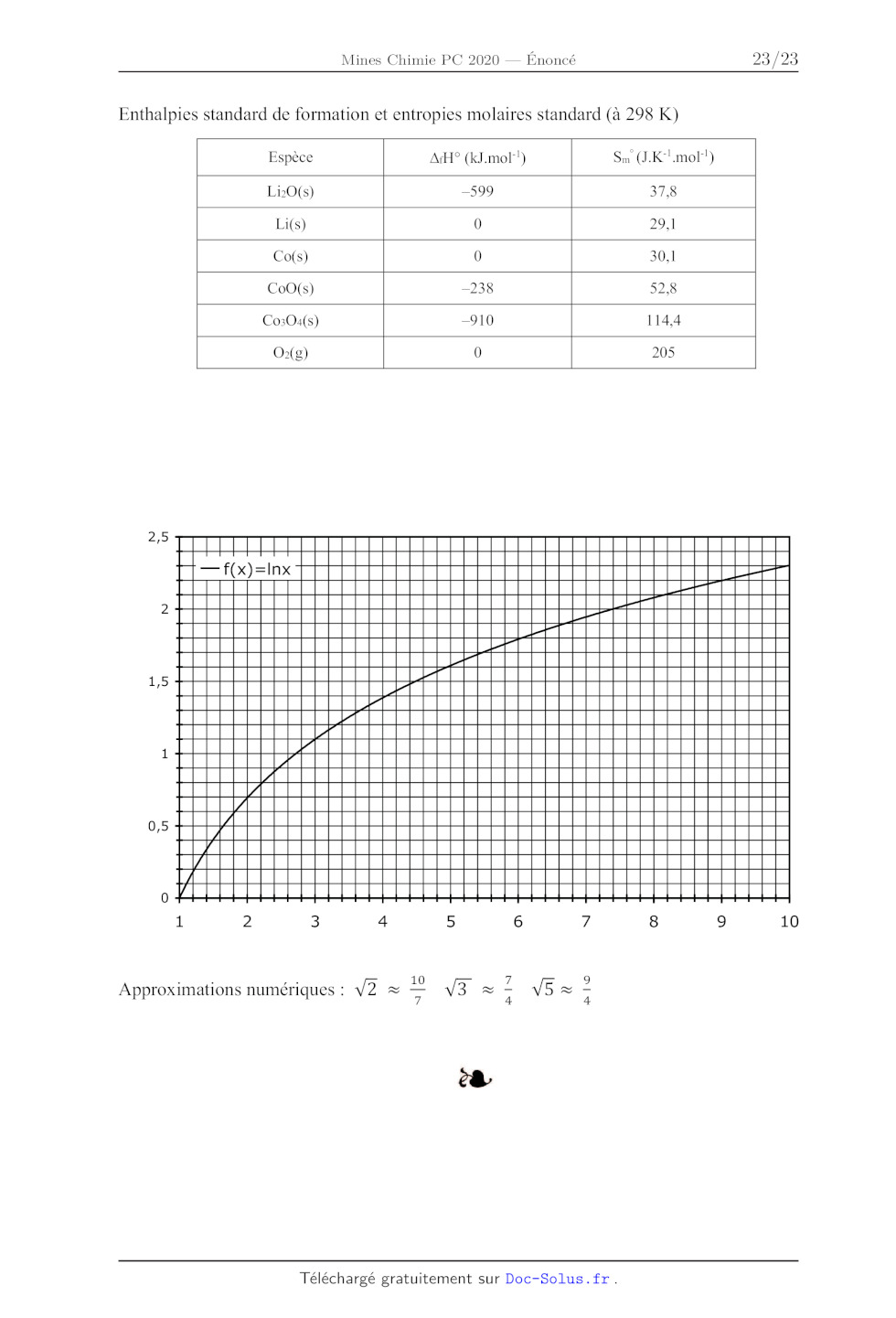
Enthalpies standard de formation et entropies molaires standard (à 298 K)
Espèce AfH° (kJ.mol!) Sm (J.K'!mol!)
LiO(s) _599 37,8
Li(s) 0 29,1
Co(s) 0 30,1
CoO(s) _238 52.8
Co3O4(s) --910 114,4
O(2) 0 205
Page 22 /22