
Centrale Chimie PC 2018
| Thème de l'épreuve | Catalyse asymétrique |
| Principaux outils utilisés | chimie organique, orbitales moléculaires, chimie de coordination, solutions aqueuses, cinétique chimique, thermodynamique |
| Mots clefs | catalyse asymétrique, complexe de Wilkinson, L-DOPA, proline, réaction de Mitsunobu, caryophyllène, enzyme, glutamate de sodium, menthone, isomenthone, Amberlyst 15 dry |
Corrigé
:👈 gratuite pour tous les corrigés si tu crées un compte
👈 gratuite pour tous les corrigés si tu crées un compte
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
👈 gratuite pour ce corrigé si tu crées un compte
- - - - - -
Énoncé complet
(télécharger le PDF)









Rapport du jury
(télécharger le PDF)


Énoncé obtenu par reconnaissance optique des caractères
t, '» C h | m le 00
3 ( 1--l
_/ PC @
cnucuuns DENTHHLE-SUPËLEE 4 heures Calculatrices autorisées N
Catalyse asymétrique
Si l'industrie pharmaceutique est régulièrement citée pour illustrer la
nécessité de développer des synthèses
énantiosélectives (en 2016, les ventes de médicaments énantiopurs devaient
atteindre 150 milliards de dollars
dans le monde, avec une croissance annuelle de 13%), les industries des
polymères, des cosmétiques et de
l'agrochimie sont également concernées.
Le monde vivant a toujours été une source d'inspiration pour les chimistes. Les
enzymes, de part leurs remar--
quables efficacité et énantiosélectivité, sont des archétypes pour la catalyse
asymétrique. Si les chimistes utilisent
souvent des catalyseurs a base de métaux ou de complexes métalliques environ la
moitié des enzymes connues
ne contiennent pas d'élément métallique dans leur site actif.
Au cours de ces dernières années, il a été établi que de petites molécules
organiques, des acides aminés notam--
ment, peuvent catalyser avec une très grande efficacité et une remarquable
énantiosélectivité, de nombreuses
transformations fondamentales en chimie fine. L'organocatalyse asymétrique
connaît actuellement un développe--
ment spectaculaire qui s'explique, non seulement par les performances atteintes
par certains organocatalyseurs,
mais aussi par la facilité de mise en oeuvre des procédés et leur bonne
adéquation avec les exigences de la chimie
verte.
Ce sujet porte sur différents types de catalyseurs utilisés dans des synthèses
énantiosélectives: métaux de
transition et complexes de métaux de transition, acides a--aminés, enzymes,
résines échangeuses d'ions.
Ce sujet comporte, en fin d'énoncé, une annexe constituée de deux documents et
de données utiles au problème.
Tout élément de réponse sera valorisé s'il est justifié et cohérent. Le
candidat pourra être amené à estimer
certaines valeurs pour parvenir aux résultats.
I Catalyse asymétrique par les métaux de transition
Historiquement, les premiers résultats marquants dans le domaine de la catalyse
asymétrique sont a replacer
dans le cadre des réactions d'hydrogénation. Des exemples de réactions
d'hydrogénation sont proposés figure 1.
0 o
1 atm H2
_
RhCI(PPh3)3
65 atm H2
_»
Raney Ni
/ ÔL
... ""
Ü ÎÛ
(CH3)2CH "(:/{GHz pt_ H2 (CHS)ZCH,_ _...CH3
OH
OH
Figure 1 Exemples de réaction d'hydrogénation
Q 1. Analyser ces exemples en commentant les conditions de réaction et les
résultats obtenus en termes de
sélectivité et d'apport dans les stratégies de synthèse.
I.A -- Orbitales moléculaires d'un compleme de géométrie plan-carré
Le complexe de Wilkinson est un des premiers complexes à avoir été utilisé pour
réaliser l'hydrogénation des
alcènes. Ce complexe, de formule [Rh(PPh3)3Cl], comprend trois ligands
triphénylphosphine et un ligand chlo--
rure.
2018-02--25 19:09:26 Page 1/12 r@EUR_
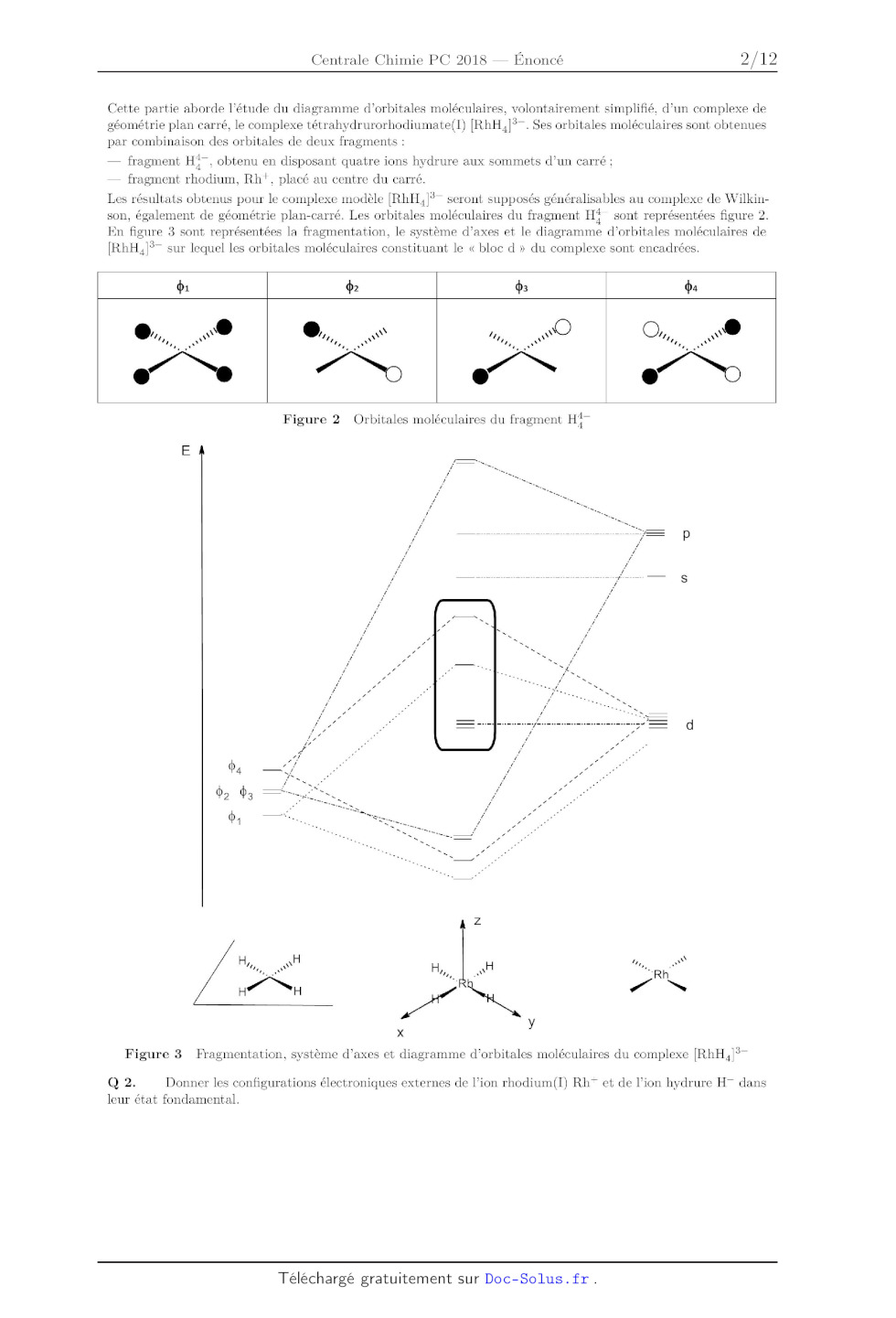
Cette partie aborde l'étude du diagramme d'orbitales moléculaires,
volontairement simplifié, d'un complexe de
géométrie plan carré, le complexe tétrahydrurorhodiumate(l) {RhH4]3Î Ses
orbitales moléculaires sont obtenues
par combinaison des orbitales de deux fragments :
* fragment HÎ, obtenu en disposant quatre ions hydrure aux sommets d'un carré ;
* fragment rhodium, Rh+, placé au centre du carré.
Les résultats obtenus pour le complexe modèle {RhH4]3* seront supposés
généralisables au complexe de Wilkin--
son, également de géométrie plan--carré. Les orbitales moléculaires du fragment
HÎÎ sont représentées figure 2.
En figure 3 sont représentées la fragmentation, le système d'axes et le
diagramme d'orbitales moléculaires de
{RhH4]3* sur lequel les orbitales moléculaires constituant le « bloc d » du
complexe sont encadrées.
du
d>z
@.
\\.
\'\
\
";
'l
I'] \
o/\o
\\
\"
'\
.,
'l
"l \
/\>
OI \\\.
I,," '\\
'\
Figure 3 Fragmentation, système d7axes et diagramme d'orbitales moléculaires du
complexe {RhH4]3*
Q2.
leur état fondamental.
2018-02--25 19:09:26
Page 2/12
Donner les configurations électroniques externes de l'ion rhodium(l) Rh+ et de
l'ion hydrure Hi dans
Q 3. En respectant le système d'axes imposé, identifier précisément avec
quelles orbitales atomiques du
rhodium, chacune des orbitales moléculaires çbl, %, (133 et @, a été combinée.
Q 4. Parmi les orbitales moléculaires du complexe, identifier, en justifiant,
une orbitale moléculaire hante,
une non--hante et une anti--hante.
Q 5. Les complexes de géométrie plan--carré sont rarement observés dans le cas
où le complexe compte 18
électrons de valence. Proposer une explication simple en vous appuyant sur le
diagramme d'orbitales molécu--
laires.
I.A.1) Extension au complexe de Wilkinson
Dans le complexe de Wilkinson, trois des ligands hydrure du complexe {RhH4]3*
sont remplacés par des ligands
triphénylphosphine dont les orbitales frontalières sont représentées
schématiquement figure 4.
E1() [3\/
Figure 4 Orbitales frontalières schématiques des ligands triphénylphosphine PPh3
Q 6. Donner le schéma de Lewis de la molécule de triphénylphosphine. Indiquer
la géométrie de la molécule
autour de l'atome central de phosphore et préciser l'ordre de grandeur des
angles entre les liaisons.
Q 7. Les ligands PPh3 sont qualifiés de a--donneurs et 7r--accepteurs.
Identifier l'orbitale frontalière mise en
jeu pour chacun de ces qualificatifs, puis schématiser l'interaction
orbitalaire illustrant les caractéristiques de
ce ligand.
I.A.2) Étude du mécanisme de l'hydr0génation
Le cycle catalytique proposé pour l'hydrogénati0n des alcènes est reproduit
figure 5.
Ph3P/Ili,, h]_\\\\Pph3
C"'R h'PPh3
E
S (Solvant)
? Etape 0
PPh3
Ph3PI/,,,] ...\S
(>er ]h'PPh3
Etape 4 Etape ]
H Ph3P/I,, ...H
T ... ...z' : »...
3
Cl/R | h'PPh3
S
4_T
Etape 3 Etape 2
Ph3P/Ülh] ,\\\\H
Cl/l ='PPh3
S
&
Figure 5 Cycle catalytique de l'hydrogènati0n des alcènes
Si le complexe de Wilkinson est stable à l'état solide, il subit, dans
l'éthanol, la substitution d'un ligand
phosphine par une molécule de solvant (notée S dans le cycle catalytique).
Lors de la première étape, le complexe 1 fixe une molécule de dihydrogëne. Afin
de modéliser l'approche des
réactifs, on suppose que la molécule de dihydr0gène approche le complexe 1 dans
le plan (yz) parallèlement à
l'axe des y, comme cela est visualisé figure 6.
2018-02--25 19:09:26 Page 3/12 Î(°°
Figure 6 Approche d'un ligand dihydrogène H2
Q 8. Écrire l'équation de la réaction associée au cycle catalytique.
Q 9. Le complexe de Wilkinson est--il le catalyseur de cette réaction ?
Justifier.
Q 10. Reconnaître la nature des étapes 1 et 3 en justifiant précisément la
réponse.
LB -- Hydrogénation énantiosélecti've en présence d'un compleme à ligands
chirauæ
La DIOP, représentée figure 7, est une diphosphine qui présente plusieurs
stéréoisomères et dont la première
application industrielle a été la synthèse de la L--Dopa (dérivé de la
phénylalanine, médicament utilisé dans le
traitement de la maladie de Parkinson) par l'équipe de Knowles (prix Nobel
2001) de la société Monsanto. Le
complexe du rhodium utilisé pour cette synthèse est {Rh(COD)LÆ*, BFZ où COD est
le cycloocta--l,5--diène et
L une phosphine chirale de type DIOP.
Me02C O
Ph2P O O
MGO2C
Figure 7 Formule topologique de la DIOP (à gauche) et d'une espèce organique de
type DIOP (à droite)
L'étape--clé de la synthèse de la L--Dopa est une hydrogénation
énantiosélective représentée figure 8. Cette étape
présente un excès énantiomérique : ee : 95%, avec ee : |OER --æS| où IR et æS
représentent les fractions molaires
respectives des énantiomères R et S.
000" COOH
/ ...... NHAc
_»
0Me OMe
OA° 0Ac
Figure 8 Étape--clé de la synthèse de la L--Dopa
Q 11. La DIOP est synthétisée a partir de l'acide tartrique (ou acide
2,3--dihydroxybutanedioi'que). Détermi--
ner le nombre de stéréoisomères de configuration que présente l'acide tartrique.
Q 12. Proposer une séquence réactionnelle utilisant des composés organiques et
minéraux et permettant
d'obtenir l'espèce organique de type DIOP représentée figure 7 à partir de
l'acide tartrique. Préciser les conditions
expérimentales permettant de réaliser ces transformations avec un bon rendement.
Q 13. Proposer une séquence réactionnelle pour obtenir la DIOP a partir du
composé organique précédent
en utilisant, entre autres, le réactif KPPh2.
Q 14. La première synthèse de la (--)--DIOP fut réalisée par l'équipe du
chimiste français H. Kagan en 1971
a partir du stéréoisomère (R,R) de l'acide tartrique. En supposant les
configurations des atomes de carbone
asymétriques inchangées, donner une représentation spatiale de la (--)--DIOP.
On expliquera soigneusement le
raisonnement.
Q 15. Déterminer le pourcentage de chaque énantiomère formé après l'étape--clé.
2018-02-25 19:09:26 Page 4/12 ÎCÔ BY--NC-SA
1
2
3
II Catalyse asymétrique par des acides a-aminés ou organocatalyse
asymétrique
Les acides a--aminés chiraux peuvent être utilisés comme inducteurs
asymétriques pour accélérer des transfor--
mations chimiques : ce procédé est qualifié « d'organocatalyse asymétrique ».
Les avantages de ce procédé sont
nombreux : les conditions réactionnelles ne sont généralement pas sensibles à
l'humidité et à l'oxygène, les acides
aminés sont facilement accessibles, de faible coût et non toxiques. L'ensemble
de ces avantages peut conférer à
ce type de réaction un bénéfice immense par rapport a la catalyse avec les
complexes de métaux de transition
pour la synthèse de composés d'intérêt médical.
II.A * À propos de la (S)-proline
La (S)--proline, représentée à droite, est l'un des 22 acides aminés
protéinogènes. Elle est Û
2) NaH
: \
OH O
5
-- Séquence 5 : obtention du caryophyllène
1) DIBAL
2) TsCl, Pyridine
_,
3) tBuOK
\o \o
6 7
M002 C
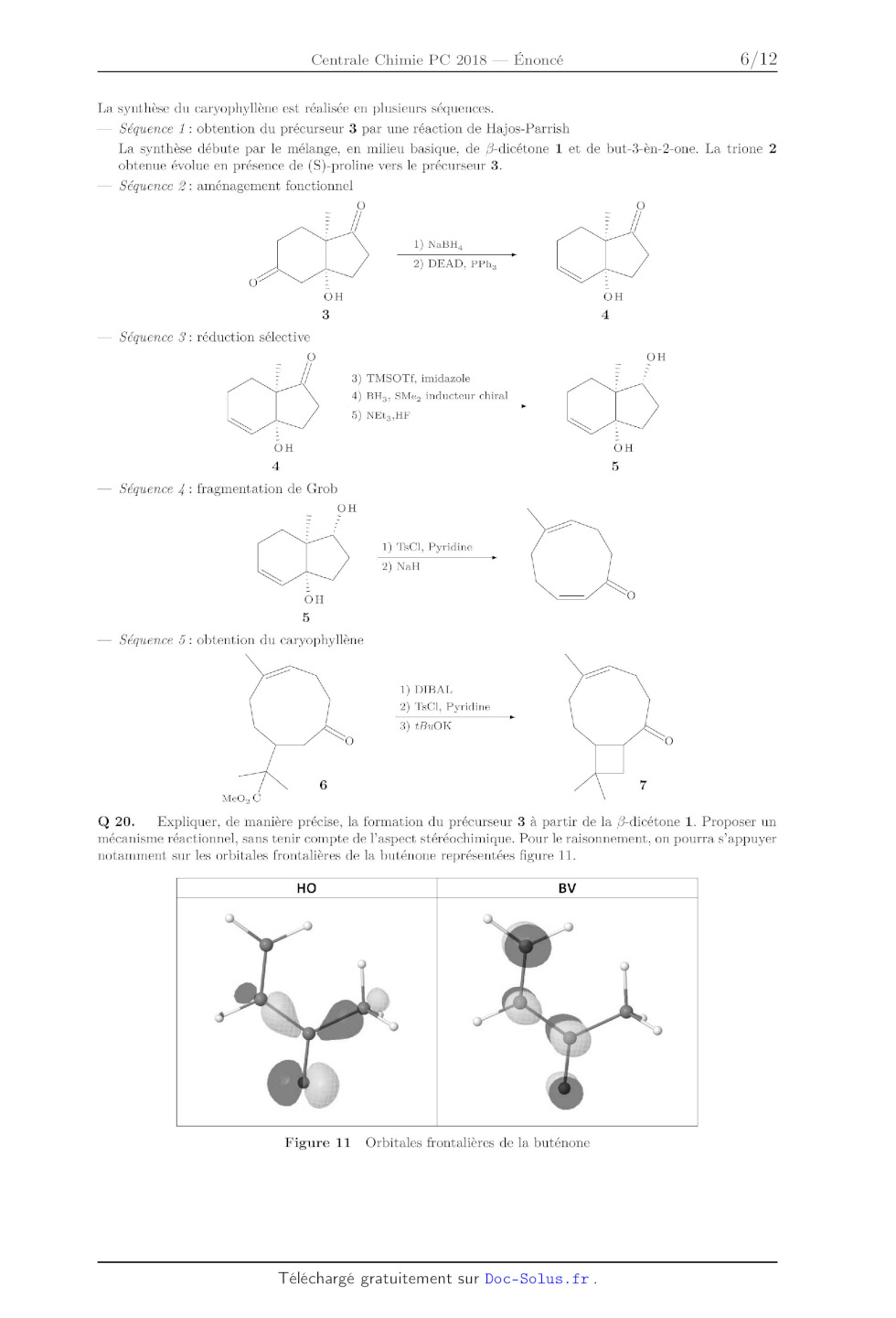
Q 20. Expliquer, de manière précise, la formation du précurseur 3 a partir de
la fl--dicétone 1. Proposer un
mécanisme réactionnel, sans tenir compte de l'aspect stéré0chimique. Pour le
raisonnement, on pourra s'appuyer
notamment sur les orbitales frontalières de la buténone représentées figure 11.
HO BV
Figure 11 Orbitales frontalières de la buténone
2018-02--25 19:09:26 Page 6/12 ÊCC BY--NC--SA
Q 21. Commenter l'appellation « organocatalyseur asymétrique » attribuée à la
(S)--proline lors de l'évolution
de 2 en 3.
Q 22. Analyser la stratégie de synthèse des deux séquences 2 et 3.
Q 23. Donner la structure du composé intermédiaire formé par action d'un
équivalent de chlorure de tosyle
sur 5, puis proposer un mécanisme pour la réaction réalisée au cours de la
seconde étape de la séquence 4.
Q 24. Expliquer la stratégie mise en place lors de la séquence 5 et commenter
les conditions expérimentales
utilisées. Comment passer du composé 7 au caryophyllène '?
III Catalyse asymétrique par des enzymes
III.A * Suivi cinétique de l'hydrolyse d'un amide catalysée par l'acylase
L'acylase extraite des reins de porcs est une enzyme capable de catalyser
l'hydrolyse du groupe amide. Son
action sur un mélange racémique de N--acétylméthionine a été étudiée par
l'équipe de Robert Olsen au moyen
de la RMN du proton 1H. Les résultats obtenus tendent à prouver que l'enzyme
n'agit que sur un seul des deux
énantiomères (figure 12).
s \
\
0 0
H |
H N
Y of Acylase Y of
_»
0 pH = 7 0
3 S
\ \
Figure 12 Action de l'acylase sur les deux énantiomères de la
N--acétylméthionine
Le protocole suivi pour cette étude expérimentale est précisé ci--dessous.
-- Solution de substrat: la solution de substrat est obtenue en diluant 192 mg
de la N--acétylméthionine racé--
mique (concentration molaire apportée Co : 0,125 molLf1 en chaque énantiomère)
et 72 mg de dihydrogé--
nophosphate de potassium, KH2PO4 (concentration molaire apportée C = 0,15
mol-L4) dans 5 mL d'eau
deutérée D20.
-- Suspension d'enzyme: la suspension d'enzyme est obtenue en introduisant 2 mg
de chlorure de cobalt(ll)
C0C12, 6H20 et 10 mg d'acylase dans 10,0 mL d'eau deutérée D20.
-- Suivi cinétique : 1,0 mL de la solution de substrat est introduit dans un
tube de RMN. La réaction débute
lors de l'introduction de 0,010 mL de la suspension d'enzyme dans le tube à
essais. Toutes les 3 minutes,
pendant environ une heure, un spectre de RMN est enregistré.
Les spectres de RMN enregistrés font apparaître deux signaux particulièrement
utiles :
-- à 4,1 ppm (intégration notée A R) pour l'atome d'hydrogène porté par l'atome
de carbone asymétrique des
réactifs non hydrolysés ;
-- a 3,7 ppm (intégration notée A P) pour l'atome d'hydrogène porté par l'atome
de carbone asymétrique du
produit d'hydrolyse.
Les résultats obtenus pour la concentration en énantiomère S de la
N--acétylméthi0nine et la vitesse de réaction 1)
au cours du temps sont consignés dans le tableau 1.
Une modélisation cinétique de la catalyse de réactions par les enzymes est
proposée dans le document 2.
Q 25. Écrire l'équation de la réaction d'hydrolyse du groupe amide dans une
solution à pH : 7.
Q 26. Expliciter les étapes à mettre en oeuvre pour calculer les données du
tableau 1 a partir des intégrations
des signaux à 3,7 et 4,1 ppm. En particulier, montrer que la concentration
{S(t)] en énantiomère S de la
N--acétylméthionine a l'instant t se calcule grâce a la relation :
lS(t)l : Com
Q 27. Déterminer, le plus précisément possible, les valeurs des paramètres
cinétiques vmax et KM caractéri--
sant l'action de l'acylase. Effectuer, au besoin, une analyse critique du
modèle utilisé.
2018-02--25 19:09:26 Page 7/12 (°_
5.0 4.5 4.0 3.5 3.0 2.5 2.0 1.5
Déplacement chimique 6 (ppm)
Figure 13 Spectre RMN 1H enregistré plusieurs minutes après le début de la
réaction
Temps (min) 6,43 10,3 13,4 16,6 19,7 22,7 25,8 28,9 31,9 35,3 38,8 41,8
(S](mmol-L") 97,6 82,8 72,9 63,5 55,4 48,1 40,5 34,1 28,4 23,5 18,3 15,0
v(mmol-L"'-min"') 3,87 3,12 3,00 2,57 2,46 2,41 2,12 1,89 1,45 1,46 1,10 0,960
Tableau 1 Concentrations en énantiomère S de la N--acétylméthionine et vitesse
de réaction
III.B -- Dosage d'un emhausteur de goût mettant en oeuvre deum enzymes
Cette sous--partie s'intéresse au dosage d'un exhausteur de goût, le glutamate
de sodium (ou glutamate md
nosodique noté GMS ou E621) dans un sachet de potage déshydraté, dosage réalisé
en mettant en oeuvre
successivement deux enzymes.
En effet, l'acide glutamique (acide (+)--(S)--2--aminopentanedioïque) est
l'acide aminé le plus abondant de l'ali--
mentation humaine. Sa saveur, différente du sucré, du salé, de l'acide et de
l'amer a été reconnue pour la
première fois en 1908 par le scientifique japonais Kikunae Ikeda, qui la nomma
« umami » (savoureux). L'acide
glutamique libre est naturellement abondant dans des fromages, les sauces soja
ou les tomates (tableau 2).
Produit Parmesan Emmental Algue Sauce soja Tomate Petit pois
Glutamate (mg/100g) 1680 308 1608 926 246 106
Tableau 2 Kumiko Ninomiya, Natural occurrence, Food Reviews International,
1998, 14, pp. 2--3
De nombreux sites internet et ouvrages présentent les «dangers» liés à la
consommation de glutamate. Ce
syndrome est associé à la consommation du GMS (provoquant des symptômes de
brûlure, d'engourdissement, des
sensations serrées dans la partie supérieure du corps) mais aucune donnée
concluante n'a pu prouver qu'il s'agit
des effets secondaires immédiatement causés par l'additif. La dose journalière
admissible (DJ A) en glutamate de
sodium (GMS) a été évaluée à 120 mg-kg"'. L'utilisation du glutamate dans les
aliments pour bébé est interdite
dans certains pays. En tant qu'additifs alimentaires, en Europe comme aux
États--Unis, les glutamates sont
exclus de la filière alimentation bio.
L'acide L--glutamique peut être dosé dans des produits commerciaux afin de
vérifier si la concentration de cet
exhausteur de goût est conforme à la règlementation. Le dosage est ici réalisé
sur une solution aqueuse de potage
déshydraté commercial (figure 14) préparée en dissolvant 1,00 g de poudre dans
1,00 L d'eau.
soupe
WP" .... -.
' ""'-"' 3 mon:
. kW'" p _
CI IlNO|Si ÆOE"OEWWÆ:ËOEHWOEQE
' ' ' W" rm (OMPASS®
"...""ü khamg'g""m "" ' . nm Fm..." Ndf'p£lEURdmts NE'Sl|(' SA.
1 d
TrJdem9:l al 5051ch
INUlSE NOUILLES, CHAMPIGNON
RATEECH ... ble : ....mn1cslËi'ï.
.* flrç
(lnulv run1"-\\nmlaln .»
Ma...... 11 ...... 1...1...--........ ... -...
Figure 14 Sachet de potage déshydraté analysé
2018-02--25 19:09:26 Page 8/12 [ËC
Le principe du dosage de l'acide L--glutamique peut être décrit de la façon
suivante.
---- En présence de l'enzyme glutamate déshydrogénase (GLDH) et de NADÏ l'acide
glutamique subit une
désamination. Cette transformation, peut être modélisée par la réaction
d'équation (1) et de constante
d'équilibre K1.
Acide L--glutamique + NAD+ + H2O : 2--oxoglutarate + NHî + NADH (1)
-- Le N ADH formé, en présence d'une seconde enzyme, la diaphorase, réduit le
chlorure d'iodonitrotétrazolium
(INT) en formazan, dont le spectre d'absorption présente un maximum a 492 nm.
Dans les conditions de
mesure, le coefficient d'absorption molaire du formazan vaut e(492 nm) : 1,99
>< 103 m2-molfl. Cette réaction d'oxyde--réduction, d'équation (2), peut être considérée comme totale (K2 >>
K1).
INT+ NADH + H+ -> NAD+ + formazan (2)
Le mode opératoire du dosage spectrophotométrique et les résultats obtenus sont
rassemblés dans le tableau 3.
Toutes les mesures d'absorbance sont effectuées en réglant le zéro du
spectrophotomètre avec de l'eau distillée,
dans des cuves dont le trajet optique vaut 1,0 cm.
Potage Témoin (eau)
Tampon (pH : 8,6) 2,50 mL
NAD+ (6,7 mmol-Lfl) 020 mL
INT (1,2 mmolL"') 020 mL
Diaphorase (15 U'mL*1) 0,050 mL
Échantillon à doser 0,10 mL de potage 0,10 mL d'eau
Mélanger et attendre 3 minutes
Mesurer l'absorbance à 492 nm A1 : 0,102 Atémoinl = 0,100
GLDH (1200 U'mL"1) 0,050 mL 0,050 mL
Mélanger et attendre 3 minutes
Mesurer l'absorbance à 492 nm A2 : 0,216 Atémoin2 : 0,105
Tableau 3 Mode opératoire et résultats obtenus lors du dosage
Q 28. Estimer le nombre de sachets de potage déshydraté qu'un adulte peut
consommer quotidiennement.
Cette question demande de l'initiative de la part du candidat. La démarche et
les pistes de recherche doivent
être consignées ,' si elles sont pertinentes, elles seront valorisées. Le
barème tient compte du temps nécessaire
pour élaborer un raisonnement, explorer éventuellement difiérentes pistes et
valider le résultat.
IV Catalyse asymétrique acido-basique
La (--)--menthone s'isomérise en (+)--isomenthone, sous l'effet d'une catalyse
acide. Plusieurs sources d'acides
peuvent être utilisées pour catalyser l'isomérisation ; cette étude porte sur
l'utilisation d'une résine échangeuse
d'ions appelée « Amberlyst 15 dry ». L'isomérisation de la (--)--menthone en
(+)--isomenthone peut être modélisée
par la réaction représentée figure 15.
(-- )--Menthone (+)-- Isomenfl10ne
Figure 15 Réaction d'isomérisation de la (--)--menthone en (+)--isomenthone
IV.A + Synthèse de la résine « Amberlyst 15 dry »
La résine échangeuse d'ions « Amberlyst 15 dry » est obtenue par traitement à
l'acide sulfurique concentré d'un
copolymère styrène--divinylbenzène dont les pourcentages massiques respectifs
de styrène et de divinylbenzène
sont de 96% et 4%.
2018-02--25 19:09:26 Page 9/12 _c)
// <_> / Ê'
? ?
H H
Figure 16 Formule du divinylbenzène et motif du polystyrène
Le traitement à l'acide sulfurique vise à fixer des groupes sulfoniques sur
certains cycles benzéniques du copoly--
mère, comme indiqué figure 17 sur l'exemple du benzène.
H + H2504 _) @ 303H + H2O
Figure 17 Sulfonation du benzène en acide benzènesulfonique
Protocole de sulfonati0n du copolymère
-- Introduire 15 mL d'acide sulfurique concentré dans un erlenmeyer de 50 mL.
Ajouter 0,02 g de sulfate d'argent
et agiter avec précaution jusqu'à dissolution complète du solide. Chauffer le
mélange à 90 °C dans un bain--
marie, puis introduire 1,0 g de billes de copolymère. Surmonter l'erlenmeyer
d'un réfrigérant et chauffer
pendant 2 heures.
-- Quand la durée est écoulée, verser précautionneusement, sur le mélange, 100
mL d'acide sulfurique concentré
froid. Filtrer sur Büchner et laver le solide avec 5 portions de 10 mL d'eau
distillée jusqu'à ce que le filtrat
soit devenu neutre.
-- Rincer le copolymère avec 2 portions de 10 mL de méthanol.
-- Recueillir les billes de résine et les sécher a l'étuve a 105 °C pendant 10
à 15 minutes.
Protocole de titrage de la résine
-- Peser 200 mg de la résine et les introduire dans 20 mL d'eau distillée.
Ajouter une goutte de phénolphtaléine
et titrer par une solution aqueuse d'hydroxyde de sodium à 0,100 mol-L". La
stabilisation de la couleur
proche de l'équivalence peut nécessiter plusieurs secondes.
-- Le changement de couleur est observé pour un volume de soude versé égal à
10,4 mL.
Q 29. Citer un intérêt d'utiliser l'Amberlyst 15 dry comme catalyseur plutôt
que l'acide sulfurique.
Q 30. Justifier l'acidité forte de l'acide benzènesulfonique H5C6 -- SO3H.
Q 31. Représenter le monomère utilisé pour former le polystyrène. Quel est
l'intérêt du divinylbenzène dans
la fabrication du copolymère '?
Q 32. Évaluer le nombre (exprimé en mol) de cycles benzéniques sulfonés par
gramme de résine.
Q 33. En déduire la valeur du rendement de la réaction de sulfonation du
copolymère.
IV.B * Suivi cinétique de l'isomérisatian de la (--)-menth0ne en (+)-isomenth0ne
La (--)--menthone et la (+)--isomenthone étant toutes deux chirales, un suivi
cinétique de la transformation est
réalisé par polarimétrie. Dans une enceinte maintenue à 65 °C, sont introduits :
-- 15 mL de (--)--menthone ;
-- 15 mL d'éthanol ;
-- 100 mg d'Amberlyst solide.
Le pouvoir rotatoire de la solution est mesuré à intervalles de temps réguliers
par prélèvement et introduction
d'un échantillon de solution dans une cuve de polarimétrie de longueur
intérieure égale à EUR = 10 cm. Les valeurs
obtenues sont regroupées dans le tableau 4.
Temps (min) 0 5 10 15 20 30 45 60 1440 1600
a(°) --10,3 --8,1 --6,0 --4,4 --2,9 --0,8 1,5 2,6 4,4 4,4
Tableau 4 Valeurs expérimentales du pouvoir rotatoire 04 en fonction du temps
Q 34. Présenter un protocole expérimental permettant de déterminer la valeur
d'un pouvoir rotatoire spéci--
fique.
Q 35. Nommer la relation d'isomérie entre la (--)--menthone et la
(+)--isomenthone. Commenter les valeurs
de leurs pouvoirs rotatoires spécifiques.
Q 36. Estimer le pourcentage de chaque isomère présent à l'état initial et à
l'état final a partir des données
expérimentales. Commenter ces résultats.
2018-02--25 19:09:26 Page 10/12 GC)--
Q 37. Soit x(t) l'avancement volumique de la réaction d'isomérisation a
l'instant t. Établir l'équation diffé--
rentielle a laquelle obéit æ(t).
Q 38. Que devient cette équation différentielle lorsque l'état d'équilibre
entre les deux isomères est atteint '?
Q 39. En déduire la valeur du rapport k1/ÏL1.
Q 40. La réaction d'isomérisation a aussi été étudiée à 111 °C et sa constante
d'équilibre K ° (t) a été dé--
terminée expérimentalement : K "(111 C'C) : 1,38. En déduire la valeur de
l'enthalpie standard de la réaction
d'isomérisation.
Annexe
DOCUMENT 1 -- Protection et déprotection des alcools par formation d'éthers
silylés
Les alcools sont facilement convertis en éthers de triméthylsilyle (TMS) par la
réaction entre un alcool et le
chlorure de triméthylsilyle (T MS--Cl) en présence d'une base faible (comme
l'imidazole ou la pyridine). Il existe
des variantes de cette réaction comme la formation d'un éther de
tert--butyldiméthylsilyle (TBDMS) ou de tert--
butyldiphénylsilyle (TBDPS) ou de triméthylsilyltrifiate (TMSOTf). L'avantage
des éthers silylés est leur inertie
vis--à--vis des bases, des nucléophiles carbonés ou azotés et des oxydants
courants. On observe que plus l'atome
de silicium est encombré, plus la réaction gagne en sélectivité des groupements
hydroxyle. Ainsi il est possible
de protéger sélectivement un alcool primaire par rapport a un alcool secondaire
en utilisant le TBDPS--Cl.
La déprotection des groupements hydroxyle protégés sous forme d'éthers silylés
est opérée de façon générale par
action des ions fiuorure Fi L'ion fiuorure est souvent apporté sous forme de
fiuorure d'ammonium ou bien de
fiuorure d'hydrogène ou encore sous forme de trifiuorure de bore.
Ph
\ | | NA \
--s'i--Cl Sii--Cl --S|i--SO2CF3 (\/NH
Ph \ N/
TMS--Cl TBDPS--Cl TMSOTf Imidazole Pyridine
DOCUMENT 2 -- Modélisation cinétique de la catalyse enzymatique
Les enzymes sont des molécules dont l'activité catalytique est directement liée
à la forme de son site actif, cavité
dans laquelle se déroule la réaction et sur laquelle ne se fixent que les
substrats de taille et de géométrie adaptées.
Les enzymes :
* accélèrent les transformations par un facteur pouvant aller jusqu'à 109 :
* agissent uniquement sur des substrats de forme adaptée au site actif :
-- sont capables d'opérer à température ambiante et a un pH proche de la
neutralité.
Le mécanisme le plus répandu pour modéliser l'action de l'enzyme E sur le
substrat S a été proposé par Leonor
Michaelis et Maud Menten en 1913 ; P désigne le produit et E--S un
intermédiaire réactionnel appelé complexe
enzyme--substrat .
k1 k
E+S ":* E-S _> E+P
f1
L'étude cinétique du mécanisme permet d'exprimer la vitesse 1; de formation du
produit P :
Uma}: ls] <=> l : K1VI + 1
v : KM + {S} " Uniafol "max
La constante K M s'exprime en fonction des constantes de vitesse k1, kil et @.
Elle caractérise l'efficacité de
la fixation du substrat sur le site actif de l'enzyme. vmax est homogène à une
vitesse ; sa valeur renseigne sur
l'efiicacité de la conversion du substrat S en produit P.
Les biochimistes expriment généralement les concentrations en enzyme au moyen
de « l'unité enzymatique »
(symbole U) qui représente la quantité d'enzyme nécessaire pour traiter une
micromole de substrat en une
minute dans des conditions opératoires (pH, température, paramètres de
solution) qui doivent être précisées.
La valeur correspond généralement aux conditions optimales pour l'activité
enzymatique, mais on normalise
parfois les valeurs à 30 °C afin de permettre les comparaisons entre enzymes.
2018-02--25 19:09:26 Page 1 1/12 'VGC_
Données
Abréviations utilisées
Groupe Méthyle Éthyle Tosyle tert--butyle Acétyle Phényle DIBAL
Formule --SOQ--CGH4--CH3 --C(CH3)3 --CO--CH3 --C6H5 /--< A] Abréviation M EUR Et Ts tBu Ac Ph < \ H Constante des gaz parfaits R = 8,31 ,l-1{*1-mol*1 pKa de différents couples acido--basiques à 25 °C -- Acide carboxylique / Carboxylate : entre 2 et 5 (exemple CH3COOH/CHBCOOÎ : 4,8) -- Ammonium/ Amine : entre 8 et 11 (exemple NHî/NH3 : 9,2) -- Acides aminés : . (S)--proline : 2,0 et 10,6 respectivement pour les couples R--COOH/R--COOÎ et R'--NHâ/R'--NH2 . Acide glutamique : 2,19 et 4,25 pour les couples R--COOH/R--COOÎ et 9,67 pour le couple R'--NHÿ/R'--NHQ Pouvoirs rotatoires spécifiques à 25 °C dans l'éthanol [cr]2D5 (° dmfl' mL. gil) TCb sous 1bar (°C) Densité à 298 K (--)--menthone --29,6 207 0,895 (+)--isomenthone +91,7 205 Masses molaires Molécule Styrène Divinylbenzène Styrène sulfoné M (g'molfl) 104 130 184 Extrait du tableau périodique des éléments Hydrogène 4-- Nom de l'élément llêlium 1 4-- Numéro atomique 2 H <-- Symbolc chimique He 1,0080 4-- Masse molaire atomique 4,0026 Lithium Béryllium Bom Carbone Azote Oxygène Fluor Néon 3 4 5 6 7 8 9 10 Li Be B C N 0 F Ne 6,9395 9,0122 10,814 12,011 14,007 15,999 18,998 20,180 Sodium Magnésium Aluminium Silicium Phosphore Soufre Chlor(z Argon 11 12 13 14 15 16 17 18 Na Mg Al Si P S Cl Ar 22,990 24,306 26,982 28,085 30,974 32,068 35,452 39,948 Potassium Calcium Scandinm Titane Vanadium Chrome Ï\Ianganèse Fer Cobalt Nickel f'nivi'e Zinc Gallium Germanium Ai'sonic Sélénium Emme Krypton 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 K Ca Sc Ti V Cr Mn Fe Co Ni Cu Zn Ga Ge As Se Br Kr 39,098 40,078 44,956 47,867 50,941 51,996 54,938 55,845 58,933 58,693 63,546 65,38 69,723 72,630 74,921 78,971 79,904 83,798 Rubidium Strontium Yttrium Zirconium l\'iobium Molybdènc T('Cllnétiuln Ruthénium R,hodium Palladium Argent Cadmium Indium Étain Antimoinc l'allure Iode Xénon 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 Rb Sr Y Zr Nb Mo Tc Ru Rh Pd Ag Cd In Sn Sb Te I Xe 85,467 87,62 88,906 91,224 92,906 95,95 [98] 101,07 102,91 106,42 107,87 112,41 114,82 118,71 121,76 127,60 126,90 131,29 ('âsiurn Bary um llafnium Tantalc 'l'ungstènc Rliénium Osmium Iridium Platine Or Mercure 'l'lmllium Plomb Bismuth Polonium Astatc Radon 55 56 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 Cs Ba Hf Ta W Re Os Ir Pt Au Hg Tl Pb Bi Po At Rn 132,91 137,33 178,49 180,948 183,84 186,21 190,23 192,22 195,08 196,97 200,59 204,38 207,2 208,98 [209] [210] [222] oooFlNooo 2018-02--25 19:09:26 Page 12/12 @_